Part 6: Hamiltonian Monte Carlo (HMC)
Introduction
This is the sixth part in a series of posts on MCMC-based Bayesian inference for a logistic regression model. If you are new to this series, please go back to Part 1.
In the previous post we saw how to construct an MCMC algorithm utilising gradient information by considering a Langevin equation having our target distribution of interest as its equilibrium. This equation has a physical interpretation in terms of the stochastic dynamics of a particle in a potential equal to minus the log of the target density. It turns out that thinking about the deterministic dynamics of a particle in such a potential can lead to more efficient MCMC algorithms.
Hamiltonian dynamics
Hamiltonian dynamics is often presented as an extension of a fairly general version of Lagrangian dynamics. However, for our purposes a rather simple version is quite sufficient, based on basic concepts from Newtonian dynamics, familiar from school. Inspired by our Langevin example, we will consider the dynamics of a particle in a potential function 
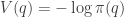
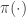


The potential function induces a (conservative) force on the particle equal to 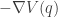

In Newtonian mechanics, we often consider the position vector 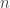
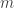
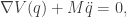
where 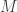
We will take the above equation as the fundamental law governing our dynamical system of interest. The motivation from Newtonian dynamics is interesting, but not required. What is important is that the dynamics of such a system are conservative, in a way that we will shortly make precise.
Our law of motion is a second-order differential equation, since it involves the second derivative of 
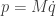
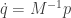
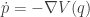
These are, in fact, Hamilton’s equations for this system, though this isn’t how they are typically written.
If we define the kinetic energy as

then the Hamiltonian
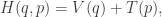
representing the total energy in the system, is conserved, since
![\displaystyle \dot{H} = \nabla V\cdot \dot{q} + \dot{p}^\text{T}M^{-1}p = \nabla V\cdot \dot{q} + \dot{p}^\text{T}\dot{q} = [\nabla V + \dot{p}]\cdot\dot{q} = 0.](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cdot%7BH%7D+%3D+%5Cnabla+V%5Ccdot+%5Cdot%7Bq%7D+%2B+%5Cdot%7Bp%7D%5E%5Ctext%7BT%7DM%5E%7B-1%7Dp+%3D+%5Cnabla+V%5Ccdot+%5Cdot%7Bq%7D+%2B+%5Cdot%7Bp%7D%5E%5Ctext%7BT%7D%5Cdot%7Bq%7D+%3D+%5B%5Cnabla+V+%2B+%5Cdot%7Bp%7D%5D%5Ccdot%5Cdot%7Bq%7D+%3D+0.+&bg=ffffff&fg=333333&s=0&c=20201002)
So, if we obey our Hamiltonian dynamics, our trajectory in 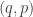
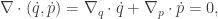
since 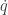
Hamiltonian Monte Carlo (HMC)
In Hamiltonian Monte Carlo we introduce an augmented target distribution,
![\displaystyle \tilde \pi(q,p) \propto \exp[-H(q,p)]](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Ctilde+%5Cpi%28q%2Cp%29+%5Cpropto+%5Cexp%5B-H%28q%2Cp%29%5D+&bg=ffffff&fg=333333&s=0&c=20201002)
It is clear from this definition that moves leaving the Hamiltonian invariant will also leave the augmented target density unchanged. By following the Hamiltonian dynamics, we will be able to make big (reversible) moves in the space that will be accepted with high probability. Also, our target factorises into two independent components as
![\displaystyle \tilde \pi(q,p) \propto \exp[-V(q)]\exp[-T(p)],](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Ctilde+%5Cpi%28q%2Cp%29+%5Cpropto+%5Cexp%5B-V%28q%29%5D%5Cexp%5B-T%28p%29%5D%2C+&bg=ffffff&fg=333333&s=0&c=20201002)
and so choosing 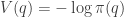
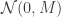

So, an idealised version of HMC would proceed as follows: First, update
Hamiltonian systems admit nice numerical integration schemes called symplectic integrators. In HMC a simple alternating Euler method is typically used, known as the leap-frog algorithm. The component updates are all shear transformations, and therefore volume preserving, and exact reversibility is ensured by starting and ending with a half-step update of the momentum variables. In principle, to ensure reversibility of the proposal the momentum variables should be sign-flipped (reversed) to finish, but in practice this doesn’t matter since it doesn’t affect the evaluation of the Hamiltonian and it will then get refreshed, anyway.
So, advancing our system by a time step 


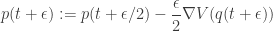
It is clear that if many such updates are chained together, adjacent momentum updates can be collapsed together, giving rise to the "leap-frog" nature of the algorithm, and therefore requiring roughly one gradient evaluation per 
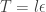
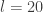
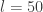
Note that since our HMC update on the augmented space consists of a Gibbs move and a M-H update, it is important that our M-H kernel does not keep or thread through the old log target density from the previous M-H update, since the Gibbs move will have changed it in the meantime.
Implementations
R
We need a M-H kernel that does not thread through the old log density.
mhKernel = function(logPost, rprop)
function(x) {
prop = rprop(x)
a = logPost(prop) - logPost(x)
if (log(runif(1)) < a)
prop
else
x
}
We can then use this to construct a M-H move as part of our HMC update.
hmcKernel = function(lpi, glpi, eps = 1e-4, l=10, dmm = 1) {
sdmm = sqrt(dmm)
leapf = function(q, p) {
p = p + 0.5*eps*glpi(q)
for (i in 1:l) {
q = q + eps*p/dmm
if (i < l)
p = p + eps*glpi(q)
else
p = p + 0.5*eps*glpi(q)
}
list(q=q, p=-p)
}
alpi = function(x)
lpi(x$q) - 0.5*sum((x$p^2)/dmm)
rprop = function(x)
leapf(x$q, x$p)
mhk = mhKernel(alpi, rprop)
function(q) {
d = length(q)
x = list(q=q, p=rnorm(d, 0, sdmm))
mhk(x)$q
}
}
See the full runnable script for further details.
Python
First a M-H kernel,
def mhKernel(lpost, rprop):
def kernel(x):
prop = rprop(x)
a = lpost(prop) - lpost(x)
if (np.log(np.random.rand()) < a):
x = prop
return x
return kernel
and then an HMC kernel.
def hmcKernel(lpi, glpi, eps = 1e-4, l=10, dmm = 1):
sdmm = np.sqrt(dmm)
def leapf(q, p):
p = p + 0.5*eps*glpi(q)
for i in range(l):
q = q + eps*p/dmm
if (i < l-1):
p = p + eps*glpi(q)
else:
p = p + 0.5*eps*glpi(q)
return (q, -p)
def alpi(x):
(q, p) = x
return lpi(q) - 0.5*np.sum((p**2)/dmm)
def rprop(x):
(q, p) = x
return leapf(q, p)
mhk = mhKernel(alpi, rprop)
def kern(q):
d = len(q)
p = np.random.randn(d)*sdmm
return mhk((q, p))[0]
return kern
See the full runnable script for further details.
JAX
Again, we want an appropriate M-H kernel,
def mhKernel(lpost, rprop, dprop = jit(lambda new, old: 1.)):
@jit
def kernel(key, x):
key0, key1 = jax.random.split(key)
prop = rprop(key0, x)
ll = lpost(x)
lp = lpost(prop)
a = lp - ll + dprop(x, prop) - dprop(prop, x)
accept = (jnp.log(jax.random.uniform(key1)) < a)
return jnp.where(accept, prop, x)
return kernel
and then an HMC kernel.
def hmcKernel(lpi, glpi, eps = 1e-4, l = 10, dmm = 1):
sdmm = jnp.sqrt(dmm)
@jit
def leapf(q, p):
p = p + 0.5*eps*glpi(q)
for i in range(l):
q = q + eps*p/dmm
if (i < l-1):
p = p + eps*glpi(q)
else:
p = p + 0.5*eps*glpi(q)
return jnp.concatenate((q, -p))
@jit
def alpi(x):
d = len(x) // 2
return lpi(x[jnp.array(range(d))]) - 0.5*jnp.sum((x[jnp.array(range(d,2*d))]**2)/dmm)
@jit
def rprop(k, x):
d = len(x) // 2
return leapf(x[jnp.array(range(d))], x[jnp.array(range(d, 2*d))])
mhk = mhKernel(alpi, rprop)
@jit
def kern(k, q):
key0, key1 = jax.random.split(k)
d = len(q)
x = jnp.concatenate((q, jax.random.normal(key0, [d])*sdmm))
return mhk(key1, x)[jnp.array(range(d))]
return kern
There is something a little bit strange about this implementation, since the proposal for the M-H move is deterministic, the function rprop just ignores the RNG key that is passed to it. We could tidy this up by making a M-H function especially for deterministic proposals. We won’t pursue this here, but this issue will crop up again later in some of the other functional languages.
See the full runnable script for further details.
Scala
A M-H kernel,
def mhKern[S](
logPost: S => Double, rprop: S => S,
dprop: (S, S) => Double = (n: S, o: S) => 1.0
): (S) => S =
val r = Uniform(0.0,1.0)
x0 =>
val x = rprop(x0)
val ll0 = logPost(x0)
val ll = logPost(x)
val a = ll - ll0 + dprop(x0, x) - dprop(x, x0)
if (math.log(r.draw()) < a) x else x0
and a HMC kernel.
def hmcKernel(lpi: DVD => Double, glpi: DVD => DVD, dmm: DVD,
eps: Double = 1e-4, l: Int = 10) =
val sdmm = sqrt(dmm)
def leapf(q: DVD, p: DVD): (DVD, DVD) =
@tailrec def go(q0: DVD, p0: DVD, l: Int): (DVD, DVD) =
val q = q0 + eps*(p0/:/dmm)
val p = if (l > 1)
p0 + eps*glpi(q)
else
p0 + 0.5*eps*glpi(q)
if (l == 1)
(q, -p)
else
go(q, p, l-1)
go(q, p + 0.5*eps*glpi(q), l)
def alpi(x: (DVD, DVD)): Double =
val (q, p) = x
lpi(q) - 0.5*sum(pow(p,2) /:/ dmm)
def rprop(x: (DVD, DVD)): (DVD, DVD) =
val (q, p) = x
leapf(q, p)
val mhk = mhKern(alpi, rprop)
(q: DVD) =>
val d = q.length
val p = sdmm map (sd => Gaussian(0,sd).draw())
mhk((q, p))._1
See the full runnable script for further details.
Haskell
A M-H kernel:
mdKernel :: (StatefulGen g m) => (s -> Double) -> (s -> s) -> g -> s -> m s
mdKernel logPost prop g x0 = do
let x = prop x0
let ll0 = logPost x0
let ll = logPost x
let a = ll - ll0
u <- (genContVar (uniformDistr 0.0 1.0)) g
let next = if ((log u) < a)
then x
else x0
return next
Note that here we are using a M-H kernel specifically for deterministic proposals, since there is no non-determinism signalled in the type signature of prop. We can then use this to construct our HMC kernel.
hmcKernel :: (StatefulGen g m) =>
(Vector Double -> Double) -> (Vector Double -> Vector Double) -> Vector Double ->
Double -> Int -> g ->
Vector Double -> m (Vector Double)
hmcKernel lpi glpi dmm eps l g = let
sdmm = cmap sqrt dmm
leapf q p = let
go q0 p0 l = let
q = q0 + (scalar eps)*p0/dmm
p = if (l > 1)
then p0 + (scalar eps)*(glpi q)
else p0 + (scalar (eps/2))*(glpi q)
in if (l == 1)
then (q, -p)
else go q p (l - 1)
in go q (p + (scalar (eps/2))*(glpi q)) l
alpi x = let
(q, p) = x
in (lpi q) - 0.5*(sumElements (p*p/dmm))
prop x = let
(q, p) = x
in leapf q p
mk = mdKernel alpi prop g
in (\q0 -> do
let d = size q0
zl <- (replicateM d . genContVar (normalDistr 0.0 1.0)) g
let z = fromList zl
let p0 = sdmm * z
(q, p) <- mk (q0, p0)
return q)
See the full runnable script for further details.
Dex
Again we can use a M-H kernel specific to deterministic proposals.
def mdKernel {s} (lpost: s -> Float) (prop: s -> s)
(x0: s) (k: Key) : s =
x = prop x0
ll0 = lpost x0
ll = lpost x
a = ll - ll0
u = rand k
select (log u < a) x x0
and use this to construct an HMC kernel.
def hmcKernel {n} (lpi: (Fin n)=>Float -> Float)
(dmm: (Fin n)=>Float) (eps: Float) (l: Nat)
(q0: (Fin n)=>Float) (k: Key) : (Fin n)=>Float =
sdmm = sqrt dmm
idmm = map (\x. 1.0/x) dmm
glpi = grad lpi
def leapf (q0: (Fin n)=>Float) (p0: (Fin n)=>Float) :
((Fin n)=>Float & (Fin n)=>Float) =
p1 = p0 + (eps/2) .* (glpi q0)
q1 = q0 + eps .* (p1*idmm)
(q, p) = apply_n l (q1, p1) \(qo, po).
pn = po + eps .* (glpi qo)
qn = qo + eps .* (pn*idmm)
(qn, pn)
pf = p + (eps/2) .* (glpi q)
(q, -pf)
def alpi (qp: ((Fin n)=>Float & (Fin n)=>Float)) : Float =
(q, p) = qp
(lpi q) - 0.5*(sum (p*p*idmm))
def prop (qp: ((Fin n)=>Float & (Fin n)=>Float)) :
((Fin n)=>Float & (Fin n)=>Float) =
(q, p) = qp
leapf q p
mk = mdKernel alpi prop
[k1, k2] = split_key k
z = randn_vec k1
p0 = sdmm * z
(q, p) = mk (q0, p0) k2
q
Note that the gradient is obtained via automatic differentiation. See the full runnable script for details.
Next steps
This was the main place that I was trying to get to when I started this series of posts. For differentiable log-posteriors (as we have in the case of Bayesian logistic regression), HMC is a pretty good algorithm for reasonably efficient posterior exploration. But there are lots of places we could go from here. We could explore the tuning of MCMC algorithms, or HMC extensions such as NUTS. We could look at MCMC algorithms that are specifically tailored to the logistic regression problem, or we could look at new MCMC algorithms for differentiable targets based on piecewise deterministic Markov processes. Alternatively, we could temporarily abandon MCMC and look at SMC or ABC approaches. Another possibility would be to abandon this multi-language approach and have a bit of a deep dive into Dex, which I think has the potential to be a great programming language for statistical computing. All of these are possibilities for the future, but I’ve a busy few weeks coming up, so the frequency of these posts is likely to substantially decrease.
Remember that all of the code associated with this series of posts is available from this github repo.
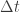 . We can improve the accuracy of this approximation by making the step size smaller, but this will come at the expense of a more slowly mixing MCMC chain.
. We can improve the accuracy of this approximation by making the step size smaller, but this will come at the expense of a more slowly mixing MCMC chain.![\displaystyle l(\beta;y) = y^\textsf{T}X\beta - \mathbf{1}^\textsf{T}\log(\mathbf{1}+\exp[X\beta])](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+l%28%5Cbeta%3By%29+%3D+y%5E%5Ctextsf%7BT%7DX%5Cbeta+-+%5Cmathbf%7B1%7D%5E%5Ctextsf%7BT%7D%5Clog%28%5Cmathbf%7B1%7D%2B%5Cexp%5BX%5Cbeta%5D%29+&bg=ffffff&fg=333333&s=0&c=20201002)
![\displaystyle l(\beta;y) = \sum_j\sum_j y_iX_{ij}\beta_j - \sum_i\log\left(1+\exp\left[\sum_jX_{ij}\beta_j\right]\right).](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+l%28%5Cbeta%3By%29+%3D+%5Csum_j%5Csum_j+y_iX_%7Bij%7D%5Cbeta_j+-+%5Csum_i%5Clog%5Cleft%281%2B%5Cexp%5Cleft%5B%5Csum_jX_%7Bij%7D%5Cbeta_j%5Cright%5D%5Cright%29.+&bg=ffffff&fg=333333&s=0&c=20201002)
 gives
gives![\displaystyle \frac{\partial l}{\partial \beta_k} = \sum_i y_iX_{ik} - \sum_i \frac{\exp\left[\sum_j X_{ij}\beta_j\right]X_{ik}}{1+\exp\left[\sum_j X_{ij}\beta_j\right]}.](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cfrac%7B%5Cpartial+l%7D%7B%5Cpartial+%5Cbeta_k%7D+%3D+%5Csum_i+y_iX_%7Bik%7D+-+%5Csum_i+%5Cfrac%7B%5Cexp%5Cleft%5B%5Csum_j+X_%7Bij%7D%5Cbeta_j%5Cright%5DX_%7Bik%7D%7D%7B1%2B%5Cexp%5Cleft%5B%5Csum_j+X_%7Bij%7D%5Cbeta_j%5Cright%5D%7D.+&bg=ffffff&fg=333333&s=0&c=20201002)
![\displaystyle \nabla l = X^\textsf{T}\left[ y - \frac{\mathbf{1}}{\mathbf{1}+\exp[-X\beta]} \right].](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cnabla+l+%3D+X%5E%5Ctextsf%7BT%7D%5Cleft%5B+y+-+%5Cfrac%7B%5Cmathbf%7B1%7D%7D%7B%5Cmathbf%7B1%7D%2B%5Cexp%5B-X%5Cbeta%5D%7D+%5Cright%5D.+&bg=ffffff&fg=333333&s=0&c=20201002)
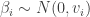 priors, it is easy to see that the gradient of the log prior is just
priors, it is easy to see that the gradient of the log prior is just  . It is the sum of these two terms that gives the gradient of the log posterior.
. It is the sum of these two terms that gives the gradient of the log posterior.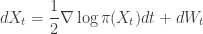

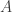 , the SDE
, the SDE
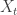 on a common scale.
on a common scale.
 is said to satisfy
is said to satisfy  if
if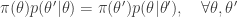
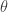 gives
gives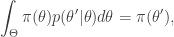
 that satisfies detailed balance. This will then give us a way to (asymptotically) generate samples from our target.
that satisfies detailed balance. This will then give us a way to (asymptotically) generate samples from our target. . Here we drop the notational dependence on
. Here we drop the notational dependence on 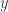 , since MCMC can be used for any target distribution of interest.
, since MCMC can be used for any target distribution of interest. and a target of interest,
and a target of interest,  to construct a new transition kernel
to construct a new transition kernel  can be described algorithmically as follows:
can be described algorithmically as follows: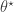 by simulating from
by simulating from  .
.![\displaystyle \alpha(\theta^\star|\theta) = \min\left[1,\frac{\pi(\theta^\star)q(\theta|\theta^\star)}{\pi(\theta)q(\theta^\star|\theta)}\right].](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Calpha%28%5Ctheta%5E%5Cstar%7C%5Ctheta%29+%3D+%5Cmin%5Cleft%5B1%2C%5Cfrac%7B%5Cpi%28%5Ctheta%5E%5Cstar%29q%28%5Ctheta%7C%5Ctheta%5E%5Cstar%29%7D%7B%5Cpi%28%5Ctheta%29q%28%5Ctheta%5E%5Cstar%7C%5Ctheta%29%7D%5Cright%5D.+&bg=ffffff&fg=333333&s=0&c=20201002)
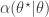 return new state
return new state  , otherwise return
, otherwise return 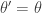 .
.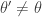 the transition kernel will be
the transition kernel will be 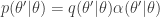 . But then
. But then![\displaystyle \pi(\theta)p(\theta'|\theta) = \min[\pi(\theta)q(\theta'|\theta),\pi(\theta')q(\theta|\theta')]](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cpi%28%5Ctheta%29p%28%5Ctheta%27%7C%5Ctheta%29+%3D+%5Cmin%5B%5Cpi%28%5Ctheta%29q%28%5Ctheta%27%7C%5Ctheta%29%2C%5Cpi%28%5Ctheta%27%29q%28%5Ctheta%7C%5Ctheta%27%29%5D+&bg=ffffff&fg=333333&s=0&c=20201002)
 , and so detailed balance is satisfied. Since detailed balance is trivial at the point mass at
, and so detailed balance is satisfied. Since detailed balance is trivial at the point mass at 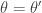 we are done.
we are done. , and so
, and so 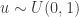 and accept if
and accept if 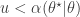 . This is convenient for several reasons. First, it means that we can ignore the "min", and just accept if
. This is convenient for several reasons. First, it means that we can ignore the "min", and just accept if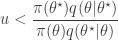
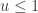 regardless. Better still, we can take logs, and accept if
regardless. Better still, we can take logs, and accept if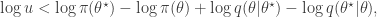
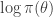 term (which in the context of Bayesian inference will typically be the log posterior) will have been computed at the previous update (irrespective of whether or not the previous move was accepted). So if we are careful about how we pass on that old value to the next iteration, we can avoid recomputing the log posterior, and our algorithm will only require one log posterior evaluation per iteration rather than two. In functional programming languages it is often convenient to pass around this current log posterior density evaluation explicitly, effectively augmenting the state space of the Markov chain to include the log posterior density.
term (which in the context of Bayesian inference will typically be the log posterior) will have been computed at the previous update (irrespective of whether or not the previous move was accepted). So if we are careful about how we pass on that old value to the next iteration, we can avoid recomputing the log posterior, and our algorithm will only require one log posterior evaluation per iteration rather than two. In functional programming languages it is often convenient to pass around this current log posterior density evaluation explicitly, effectively augmenting the state space of the Markov chain to include the log posterior density.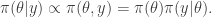
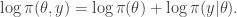
 . We assumed independent mean zero normal priors for the components of this vector, so the log prior is just the sum of logs of normal densities. Many scientific libraries will have built-in functions for returning the log-pdf of standard distributions, but if an explicit form is required, the log of the density of a
. We assumed independent mean zero normal priors for the components of this vector, so the log prior is just the sum of logs of normal densities. Many scientific libraries will have built-in functions for returning the log-pdf of standard distributions, but if an explicit form is required, the log of the density of a 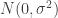 at
at 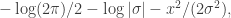
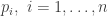 , the likelihood associated with observation
, the likelihood associated with observation  is
is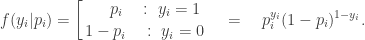
 we have
we have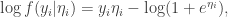

 is the linear predictor, so
is the linear predictor, so  , giving
, giving![\displaystyle \ell(\beta;y) = y^\textsf{T}X\beta - \mathbf{1}^\textsf{T}\log(\mathbf{1}+\exp[X\beta]).](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cell%28%5Cbeta%3By%29+%3D+y%5E%5Ctextsf%7BT%7DX%5Cbeta+-+%5Cmathbf%7B1%7D%5E%5Ctextsf%7BT%7D%5Clog%28%5Cmathbf%7B1%7D%2B%5Cexp%5BX%5Cbeta%5D%29.+&bg=ffffff&fg=333333&s=0&c=20201002)
 is 1 for success and -1 for failure. This isn’t really a problem, since the two encodings are related by
is 1 for success and -1 for failure. This isn’t really a problem, since the two encodings are related by  . This new encoding is convenient in the context of a logit parameterisation since then
. This new encoding is convenient in the context of a logit parameterisation since then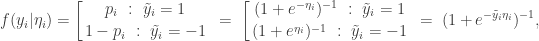

![\displaystyle \ell(\eta;\tilde y) = -\mathbf{1}\cdot \log(\mathbf{1}+\exp[-\tilde y\circ \eta]),](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cell%28%5Ceta%3B%5Ctilde+y%29+%3D+-%5Cmathbf%7B1%7D%5Ccdot+%5Clog%28%5Cmathbf%7B1%7D%2B%5Cexp%5B-%5Ctilde+y%5Ccirc+%5Ceta%5D%29%2C+&bg=ffffff&fg=333333&s=0&c=20201002)
 denotes the
denotes the ![\displaystyle \ell(\beta;\tilde y) = -\mathbf{1}^\textsf{T} \log(\mathbf{1}+\exp[-\tilde y\circ X\beta]).](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cell%28%5Cbeta%3B%5Ctilde+y%29+%3D+-%5Cmathbf%7B1%7D%5E%5Ctextsf%7BT%7D+%5Clog%28%5Cmathbf%7B1%7D%2B%5Cexp%5B-%5Ctilde+y%5Ccirc+X%5Cbeta%5D%29.+&bg=ffffff&fg=333333&s=0&c=20201002)
![\displaystyle \ell(\beta; y) = -\mathbf{1}^\textsf{T} \log(\mathbf{1}+\exp[-(2y-\mathbf{1})\circ (X\beta)]),](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cell%28%5Cbeta%3B+y%29+%3D+-%5Cmathbf%7B1%7D%5E%5Ctextsf%7BT%7D+%5Clog%28%5Cmathbf%7B1%7D%2B%5Cexp%5B-%282y-%5Cmathbf%7B1%7D%29%5Ccirc+%28X%5Cbeta%29%5D%29%2C+&bg=ffffff&fg=333333&s=0&c=20201002)
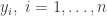 are 1 for a "success" and 0 for a "failure". The covariate
are 1 for a "success" and 0 for a "failure". The covariate 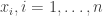 all have 1 as the first element. The statistical model is
all have 1 as the first element. The statistical model is![\displaystyle \text{logit}\left(\text{Pr}[Y_i = 1]\right) = x_i \cdot \beta,\quad i=1,\ldots,n,](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Ctext%7Blogit%7D%5Cleft%28%5Ctext%7BPr%7D%5BY_i+%3D+1%5D%5Cright%29+%3D+x_i+%5Ccdot+%5Cbeta%2C%5Cquad+i%3D1%2C%5Cldots%2Cn%2C+&bg=ffffff&fg=333333&s=0&c=20201002)
 , and
, and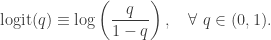
![\displaystyle \text{Pr}[Y_i = 1] = \text{expit}(x_i \cdot \beta),\quad i=1,\ldots,n,](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Ctext%7BPr%7D%5BY_i+%3D+1%5D+%3D+%5Ctext%7Bexpit%7D%28x_i+%5Ccdot+%5Cbeta%29%2C%5Cquad+i%3D1%2C%5Cldots%2Cn%2C+&bg=ffffff&fg=333333&s=0&c=20201002)
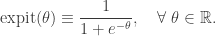
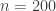 observations and 7 predictors. Adding an intercept gives
observations and 7 predictors. Adding an intercept gives  covariates. For the Bayesian analysis, we need a prior on
covariates. For the Bayesian analysis, we need a prior on