Introduction
There is a fairly large literature on reaction-diffusion modelling using partial differential equations (PDEs). There is also a fairly large literature on stochastic modelling of coupled chemical reactions, which account for the discreteness of reacting species at low concentrations. There is some literature on combining the two, to form stochastic reaction-diffusion systems, but much less.
In this post we will look at one approach to the stochastic reaction-diffusion problem, based on an underlying stochastic process often described by the reaction diffusion master equation (RDME). We will start by generating exact realisations from this process using the spatial Gillespie algorithm, before switching to a continuous stochastic approximation often known as the spatial chemical Langevin equation (spatial CLE). For fine discretisations, this spatial CLE is just an explicit numerical scheme for an associated reaction-diffusion stochastic partial differential equation (SPDE), and we can easily contrast such SPDE dynamics with their deterministic PDE approximation. We will investigate using simulation, based on my Scala library, scala-smfsb, which accompanies the third edition of my textbook, Stochastic modelling for systems biology, as discussed in previous posts.
All of the code used to generate the plots and movies in this post is available in my blog repo, and is very simple to build and run.
The Lotka-Volterra reaction network
Exact simulation from the RDME
My favourite toy coupled chemical reaction network is the Lotka-Volterra predator-prey system, presented as the three reactions
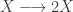
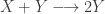
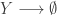
with 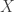
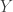
val r = 100; val c = 120 val model = SpnModels.lv[IntState]() val step = Spatial.gillespie2d(model, DenseVector(0.6, 0.6), maxH=1e12) val x00 = DenseVector(0, 0) val x0 = DenseVector(50, 100) val xx00 = PMatrix(r, c, Vector.fill(r*c)(x00)) val xx0 = xx00.updated(c/2, r/2, x0) val s = Stream.iterate(xx0)(step(_,0.0,0.1))
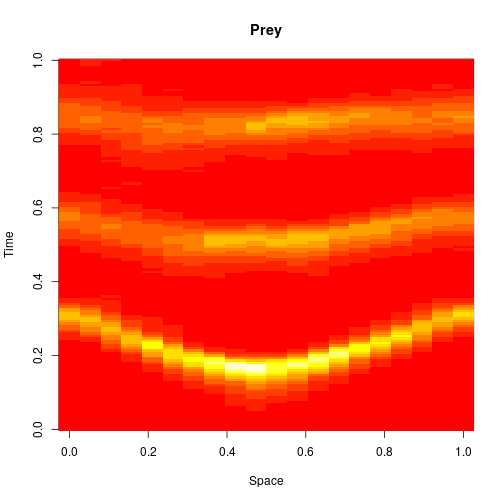
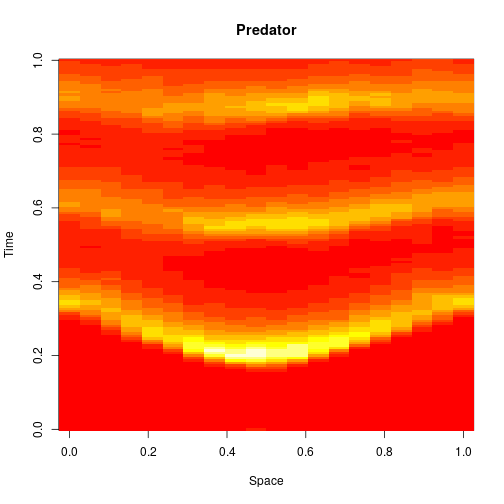
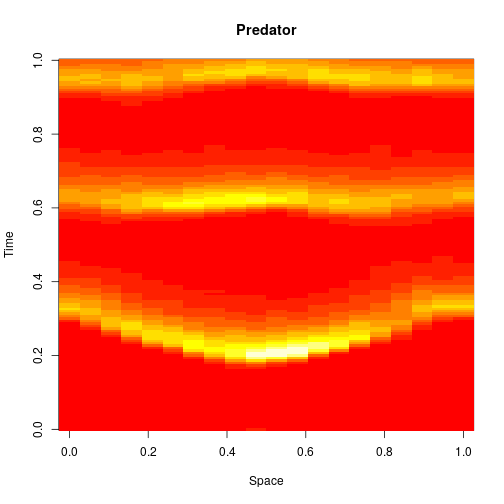
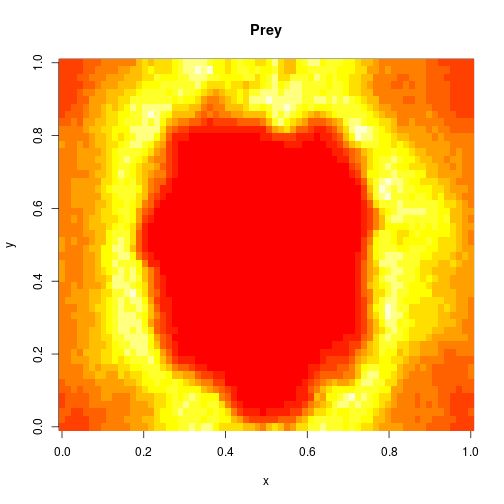
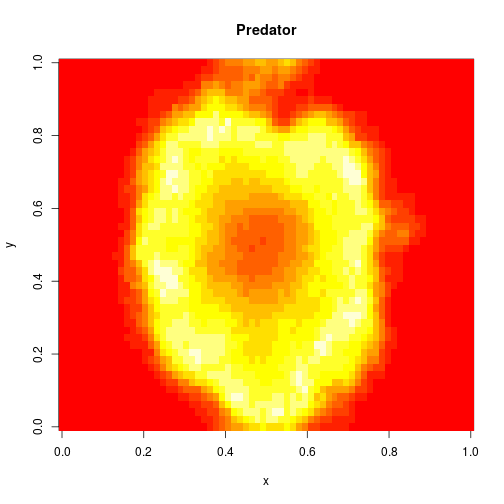
which sets up an infinite lazy Stream of states on a 100×120 grid over time steps of 0.1 units with diffusion rates of 0.6 for both species. We can then map this to a stream of images and visualise it using my scala-view library (described in this post). Running gives the following output:

The above image is the final frame of a movie which can be viewed by clicking on the image. In the simulation, blue represents the prey species, Spatial.gillespie2d in the scala-smfsb library) is quite inefficient. A more efficient implementation would use the next subvolume method or similar algorithm. But since every reaction event is simulated sequentially, this algorithm is always going to be intolerably slow for most interesting problems.
The spatial CLE
The spatial CLE effectively approximates the true RDME dynamics with a set of coupled stochastic differential equations (SDEs) on the spatial grid. This can be interpreted as an explicit scheme for numerically integrating an SPDE. But this numerical scheme is much more efficient, allowing sensible time-stepping of the process, and vectorises and parallelises nicely. The details are in my book, but the Scala implementation is here. Diffusion is implemented efficiently and in parallel using the comonadic approach that I’ve described previously. We can quickly and easily generate large simulations using the spatial CLE. Here is a movie generated on a 250×300 grid.

Again, clicking on the frame should give the movie. We see that although the quantitative details are slightly different to the exact algorithm, the essential qualitative behaviour of the system is captured well by the spatial CLE. Full code for this simulation is here.
Reaction-diffusion PDE
If we remove all of the noise terms from the spatial CLE, we get a set of coupled ODEs, which again, may be interpreted as a numerical scheme for integrating a reaction-diffusion PDE model. Below are the dynamics on the same 250×300 grid.

It seems a bit harsh to describe a reaction-diffusion PDE as “boring”, but it certainly isn’t as interesting as the stochastic dynamics. Also, it has qualitatively quite different behaviour to the stochastic models, with wavefronts being less pronounced and less well separated. The code for this one is here.
Other initialisations
Instead of just seeding the simulation with some individuals in the central pixel, we can initialise 3 pixels. We can look first at a spatial CLE simulation.
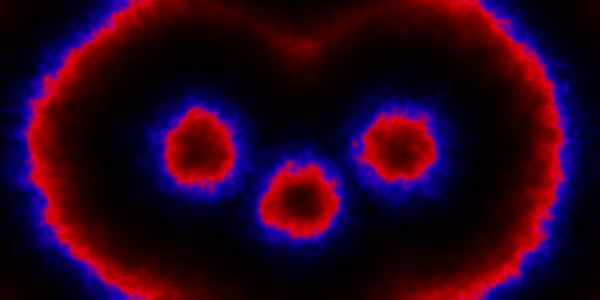
Code here.
We can look at the same problem, but now using a PDE.

Code here.
Alternatively, we can initialise every pixel independently with random numbers of predator and prey. A movie for this is given below, following a short warm-up.

Code here.
Again, we can look at the corresponding deterministic integration.

Code here.
The SIR model
Let’s now turn attention to a spatial epidemic process model, the spatial susceptible-infectious-recovered model. Again, we’ll start from the discrete reaction formulation.

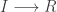
I’ll add this model to the next release of scala-smfsb, but in the meantime we can easily define it ourselves with:
def sir[S: State](p: DenseVector[Double] = DenseVector(0.1, 0.5)): Spn[S] =
UnmarkedSpn[S](
List("S", "I", "R"),
DenseMatrix((1, 1, 0), (0, 1, 0)),
DenseMatrix((0, 2, 0), (0, 0, 1)),
(x, t) => {
val xd = x.toDvd
DenseVector(
xd(0) * xd(1) * p(0), xd(1) * p(1)
)}
)
We can seed a simulation with a few infectious individuals in the centre of a roughly homogeneous population of susceptibles.
Spatial CLE
This time we’ll skip the exact simulation, since it’s very slow, and go straight to the spatial CLE. A simulation on a 250×300 grid is given below.

Here, green represents 

Code here.
PDE model
If we ditch the noise to get a reaction-diffusion PDE model, the dynamics are as follows.

Again, we see that the deterministic model is quite different to the stochastic version, and kind-of boring. Code here.
Further information
All of the code used to generate the plots and movies in this post is available in an easily runnable form in my blog repo. It is very easy to adapt the examples to vary parameters and initial conditions, and to study other reaction systems. Further details relating to stochastic reaction-diffusion modelling based on the RDME can be found in Chapter 9 of my textbook, Stochastic modelling for systems biology, third edition.