Background
In May I attended a great workshop on advances and challenges in machine learning languages at the CMS in Cambridge. There was an a good mix of people from different disciplines, and a bit of a theme around probabilistic programming. The workshop schedule includes links to many of the presentations, and is generally worth browsing. In particular, it includes a link to the slides for my presentation on a compositional approach to scalable Bayesian computation and probabilistic programming. I’ve given a few talks on this kind of thing over the last couple of years, at Newcastle, at the Isaac Newton Institute in Cambridge (twice), and at CIRM in France. But I think I explained things best at this workshop at the CMS, though my impression could partly have been a reflection of the more interested and relevant audience. In the talk I started with a basic explanation of why ideas from category theory and functional programming can help to solve problems in statistical computing in a more composable and scalable way, before moving on to discuss probability monads and their fundamental connection to probabilistic programming. The take home message from the talk is that if you have a generic inference algorithm, expressing the logic in the context of probability monads can give you an embedded probabilistic programming language (PPL) for that inference algorithm essentially “for free”.
So, during my talk I said something a little fool-hardy. I can’t remember my exact words, but while presenting the idea behind an SMC-based probability monad I said something along the lines of “one day I will write a blog post on how to write a probabilistic programming language from scratch in 50 lines of code, and this is how I’ll do it“! Rather predictably (with hindsight), immediately after my talk about half a dozen people all pleaded with me to urgently write the post! I’ve been a little busy since then, but now that things have settled down a little for the summer, I’ve some time to think and code, so here is that post.
Introduction
The idea behind this post is to show that, if you think about the problem in the right way, and use a programming language with syntactic support for monadic composition, then producing a flexible, general, compositional, embedded domain specific language (DSL) for probabilistic programming based on a given generic inference algorithm is no more effort than hard-coding two or three illustrative examples. You would need to code up two or three examples for a paper anyway, but providing a PPL is way more useful. There is also an interesting converse to this, which is that if you can’t easily produce a PPL for your “general” inference algorithm, then perhaps it isn’t quite as “general” as you thought. I’ll try to resist exploring that here…
To illustrate these principles I want to develop a fairly minimal PPL, so that the complexities of the inference algorithm don’t hide the simplicity of the PPL embedding. Importance sampling with resampling is probably the simplest useful generic Bayesian inference algorithm to implement, so that’s what I’ll use. Note that there are many limitations of the approach that I will adopt, which will make it completely unsuitable for “real” problems. In particular, this implementation is: inefficient, in terms of both compute time and memory usage, statistically inefficient for deep nesting and repeated conditioning, due to the particle degeneracy problem, specific to a particular probability monad, strictly evaluated, impure (due to mutation of global random number state), etc. All of these things are easily fixed, but all at the expense of greater abstraction, complexity and lines of code. I’ll probably discuss some of these generalisations and improvements in future posts, but for this post I want to keep everything as short and simple as practical. It’s also worth mentioning that there is nothing particularly original here. Many people have written about monadic embedded PPLs, and several have used an SMC-based monad for illustration. I’ll give some pointers to useful further reading at the end.
The language, in 50 lines of code
Without further ado, let’s just write the PPL. I’m using plain Scala, with just a dependency on the Breeze scientific library, which I’m going to use for simulating random numbers from standard distributions, and evaluation of their log densities. I have a directory of materials associated with this post in a git repo. This post is derived from an executable tut document (so you know it works), which can be found here. If you just want to follow along copying code at the command prompt, just run sbt from an empty or temp directory, and copy the following to spin up a Scala console with the Breeze dependency:
set libraryDependencies += "org.scalanlp" %% "breeze" % "1.0-RC4"
set libraryDependencies += "org.scalanlp" %% "breeze-natives" % "1.0-RC4"
set scalaVersion := "2.13.0"
console
We start with a couple of Breeze imports
import breeze.stats.{distributions => bdist}
import breeze.linalg.DenseVector
which are not strictly necessary, but clean up the subsequent code. We are going to use a set of weighted particles to represent a probability distribution empirically, so we’ll start by defining an appropriate ADT for these:
implicit val numParticles = 300
case class Particle[T](v: T, lw: Double) { // value and log-weight
def map[S](f: T => S): Particle[S] = Particle(f(v), lw)
}
We also include a map method for pushing a particle through a transformation, and a default number of particles for sampling and resampling. 300 particles are enough for illustrative purposes. Ideally it would be good to increase this for more realistic experiments. We can use this particle type to build our main probability monad as follows.
trait Prob[T] {
val particles: Vector[Particle[T]]
def map[S](f: T => S): Prob[S] = Empirical(particles map (_ map f))
def flatMap[S](f: T => Prob[S]): Prob[S] = {
Empirical((particles map (p => {
f(p.v).particles.map(psi => Particle(psi.v, p.lw + psi.lw))
})).flatten).resample
}
def resample(implicit N: Int): Prob[T] = {
val lw = particles map (_.lw)
val mx = lw reduce (math.max(_,_))
val rw = lw map (lwi => math.exp(lwi - mx))
val law = mx + math.log(rw.sum/(rw.length))
val ind = bdist.Multinomial(DenseVector(rw.toArray)).sample(N)
val newParticles = ind map (i => particles(i))
Empirical(newParticles.toVector map (pi => Particle(pi.v, law)))
}
def cond(ll: T => Double): Prob[T] =
Empirical(particles map (p => Particle(p.v, p.lw + ll(p.v))))
def empirical: Vector[T] = resample.particles.map(_.v)
}
case class Empirical[T](particles: Vector[Particle[T]]) extends Prob[T]
Note that if you are pasting into the Scala REPL you will need to use :paste mode for this. So Prob[_] is our base probability monad trait, and Empirical[_] is our simplest implementation, which is just a collection of weighted particles. The method flatMap forms the naive product of empirical measures and then resamples in order to stop an explosion in the number of particles. There are two things worth noting about the resample method. The first is that the log-sum-exp trick is being used to avoid overflow and underflow when the log weights are exponentiated. The second is that although the method returns an equally weighted set of particles, the log weights are all set in order that the average raw weight of the output set matches the average raw weight of the input set. This is a little tricky to explain, but it turns out to be necessary in order to correctly propagate conditioning information back through multiple monadic binds (flatMaps). The cond method allows conditioning of a distribution using an arbitrary log-likelihood. It is included for comparison with some other implementations I will refer to later, but we won’t actually be using it, so we could save two lines of code here if necessary. The empirical method just extracts an unweighted set of values from a distribution for subsequent analysis.
It will be handy to have a function to turn a bunch of unweighted particles into a set of particles with equal weights (a sort-of inverse of the empirical method just described), so we can define that as follows.
def unweighted[T](ts: Vector[T], lw: Double = 0.0): Prob[T] =
Empirical(ts map (Particle(_, lw)))
Probabilistic programming is essentially trivial if we only care about forward sampling. But interesting PPLs allow us to condition on observed values of random variables. In the context of SMC, this is simplest when the distribution being conditioned has a tractable log-likelihood. So we can now define an extension of our probability monad for distributions with a tractable log-likelihood, and define a bunch of convenient conditioning (or “fitting”) methods using it.
trait Dist[T] extends Prob[T] {
def ll(obs: T): Double
def ll(obs: Seq[T]): Double = obs map (ll) reduce (_+_)
def fit(obs: Seq[T]): Prob[T] =
Empirical(particles map (p => Particle(p.v, p.lw + ll(obs))))
def fitQ(obs: Seq[T]): Prob[T] = Empirical(Vector(Particle(obs.head, ll(obs))))
def fit(obs: T): Prob[T] = fit(List(obs))
def fitQ(obs: T): Prob[T] = fitQ(List(obs))
}
The only unimplemented method is ll(). The fit method re-weights a particle set according to the observed log-likelihood. For convenience, it also returns a particle cloud representing the posterior-predictive distribution of an iid value from the same distribution. This is handy, but comes at the expense of introducing an additional particle cloud. So, if you aren’t interested in the posterior predictive, you can avoid this cost by using the fitQ method (for “fit quick”), which doesn’t return anything useful. We’ll see examples of this in practice, shortly. Note that the fitQ methods aren’t strictly required for our “minimal” PPL, so we can save a couple of lines by omitting them if necessary. Similarly for the variants which allow conditioning on a collection of iid observations from the same distribution.
At this point we are essentially done. But for convenience, we can define a few standard distributions to help get new users of our PPL started. Of course, since the PPL is embedded, it is trivial to add our own additional distributions later.
case class Normal(mu: Double, v: Double)(implicit N: Int) extends Dist[Double] {
lazy val particles = unweighted(bdist.Gaussian(mu, math.sqrt(v)).sample(N).toVector).particles
def ll(obs: Double) = bdist.Gaussian(mu, math.sqrt(v)).logPdf(obs) }
case class Gamma(a: Double, b: Double)(implicit N: Int) extends Dist[Double] {
lazy val particles = unweighted(bdist.Gamma(a, 1.0/b).sample(N).toVector).particles
def ll(obs: Double) = bdist.Gamma(a, 1.0/b).logPdf(obs) }
case class Poisson(mu: Double)(implicit N: Int) extends Dist[Int] {
lazy val particles = unweighted(bdist.Poisson(mu).sample(N).toVector).particles
def ll(obs: Int) = bdist.Poisson(mu).logProbabilityOf(obs) }
Note that I’ve parameterised the Normal and Gamma the way that statisticians usually do, and not the way they are usually parameterised in scientific computing libraries (such as Breeze).
That’s it! This is a complete, general-purpose, composable, monadic PPL, in 50 (actually, 48, and fewer still if you discount trailing braces) lines of code. Let’s now see how it works in practice.
Examples
Normal random sample
We’ll start off with just about the simplest slightly interesting example I can think of: Bayesian inference for the mean and variance of a normal distribution from a random sample.
import breeze.stats.{meanAndVariance => meanVar}
// import breeze.stats.{meanAndVariance=>meanVar}
val mod = for {
mu <- Normal(0, 100)
tau <- Gamma(1, 0.1)
_ <- Normal(mu, 1.0/tau).fitQ(List(8.0,9,7,7,8,10))
} yield (mu,tau)
// mod: Wrapped.Prob[(Double, Double)] = Empirical(Vector(Particle((8.718127116254472,0.93059589932682),-15.21683812389373), Particle((7.977706390420308,1.1575288208065433),-15.21683812389373), Particle((7.977706390420308,1.1744750937611985),-15.21683812389373), Particle((7.328100552769214,1.1181787982959164),-15.21683812389373), Particle((7.977706390420308,0.8283737237370494),-15.21683812389373), Particle((8.592847414557049,2.2934836446009026),-15.21683812389373), Particle((8.718127116254472,1.498741032928539),-15.21683812389373), Particle((8.592847414557049,0.2506065368748732),-15.21683812389373), Particle((8.543283880264225,1.127386759627675),-15.21683812389373), Particle((7.977706390420308,1.3508728798704925),-15.21683812389373), Particle((7.977706390420308,1.1134430556990933),-15.2168...
val modEmp = mod.empirical
// modEmp: Vector[(Double, Double)] = Vector((7.977706390420308,0.8748006833362748), (6.292345096890432,0.20108091703626174), (9.15330820843396,0.7654238730107492), (8.960935105658741,1.027712984079369), (7.455292602273359,0.49495749079351836), (6.911716909394562,0.7739749058662421), (6.911716909394562,0.6353785792877397), (7.977706390420308,1.1744750937611985), (7.977706390420308,1.1134430556990933), (8.718127116254472,1.166399872049532), (8.763777227034538,1.0468304705769353), (8.718127116254472,0.93059589932682), (7.328100552769214,1.6166695922250236), (8.543283880264225,0.4689300351248357), (8.543283880264225,2.0028918490755094), (7.536025958690963,0.6282318170458533), (7.328100552769214,1.6166695922250236), (7.049843463553113,0.20149378088848635), (7.536025958690963,2.3565657669819897...
meanVar(modEmp map (_._1)) // mu
// res0: breeze.stats.meanAndVariance.MeanAndVariance = MeanAndVariance(8.311171010932343,0.4617800639333532,300)
meanVar(modEmp map (_._2)) // tau
// res1: breeze.stats.meanAndVariance.MeanAndVariance = MeanAndVariance(0.940762723934599,0.23641881704888842,300)
Note the use of the empirical method to turn the distribution into an unweighted set of particles for Monte Carlo analysis. Anyway, the main point is that the syntactic sugar for monadic binds (flatMaps) provided by Scala’s for-expressions (similar to do-notation in Haskell) leads to readable code not so different to that in well-known general-purpose PPLs such as BUGS, JAGS, or Stan. There are some important differences, however. In particular, the embedded DSL has probabilistic programs as regular values in the host language. These may be manipulated and composed like other values. This makes this probabilistic programming language more composable than the aforementioned languages, which makes it much simpler to build large, complex probabilistic programs from simpler, well-tested, components, in a scalable way. That is, this PPL we have obtained “for free” is actually in many ways better than most well-known PPLs.
Noisy measurements of a count
Here we’ll look at the problem of inference for a discrete count given some noisy iid continuous measurements of it.
val mod = for {
count <- Poisson(10)
tau <- Gamma(1, 0.1)
_ <- Normal(count, 1.0/tau).fitQ(List(4.2,5.1,4.6,3.3,4.7,5.3))
} yield (count, tau)
// mod: Wrapped.Prob[(Int, Double)] = Empirical(Vector(Particle((5,4.488795220669575),-11.591037521513753), Particle((5,1.7792314573063672),-11.591037521513753), Particle((5,2.5238021156137673),-11.591037521513753), Particle((4,3.280754333896923),-11.591037521513753), Particle((5,2.768438569482849),-11.591037521513753), Particle((4,1.3399975573518912),-11.591037521513753), Particle((5,1.1792835858615431),-11.591037521513753), Particle((5,1.989491156206883),-11.591037521513753), Particle((4,0.7825254987152054),-11.591037521513753), Particle((5,2.7113936834028793),-11.591037521513753), Particle((5,3.7615196800240387),-11.591037521513753), Particle((4,1.6833300961124709),-11.591037521513753), Particle((5,2.749183220798113),-11.591037521513753), Particle((5,2.1074062883430202),-11.591037521513...
val modEmp = mod.empirical
// modEmp: Vector[(Int, Double)] = Vector((4,3.243786594839479), (4,1.5090869158886693), (4,1.280656912383482), (5,2.0616356908358195), (5,3.475433097869503), (5,1.887582611202514), (5,2.8268877720514745), (5,0.9193261688050818), (4,1.7063629502805908), (5,2.116414832864841), (5,3.775508828984636), (5,2.6774941123762814), (5,2.937859946593459), (5,1.2047689975166402), (5,2.5658806161572656), (5,1.925890364268593), (4,1.0194093176888832), (5,1.883288825936725), (5,4.9503779454422965), (5,0.9045613180858916), (4,1.5795027943928661), (5,1.925890364268593), (5,2.198539449287062), (5,1.791363956348445), (5,0.9853760689818026), (4,1.6541388923071607), (5,2.599899960899971), (4,1.8904423810277957), (5,3.8983183765907836), (5,1.9242319515895554), (5,2.8268877720514745), (4,1.772120802027519), (5,2...
meanVar(modEmp map (_._1.toDouble)) // count
// res2: breeze.stats.meanAndVariance.MeanAndVariance = MeanAndVariance(4.670000000000004,0.23521739130434777,300)
meanVar(modEmp map (_._2)) // tau
// res3: breeze.stats.meanAndVariance.MeanAndVariance = MeanAndVariance(1.9678279101913874,0.9603971613375548,300)
I’ve included this mainly as an example of inference for a discrete-valued parameter. There are people out there who will tell you that discrete parameters are bad/evil/impossible. This isn’t true – discrete parameters are cool!
Linear model
Because our PPL is embedded, we can take full advantage of the power of the host programming language to build our models. Let’s explore this in the context of Bayesian estimation of a linear model. We’ll start with some data.
val x = List(1.0,2,3,4,5,6)
// x: List[Double] = List(1.0, 2.0, 3.0, 4.0, 5.0, 6.0)
val y = List(3.0,2,4,5,5,6)
// y: List[Double] = List(3.0, 2.0, 4.0, 5.0, 5.0, 6.0)
val xy = x zip y
// xy: List[(Double, Double)] = List((1.0,3.0), (2.0,2.0), (3.0,4.0), (4.0,5.0), (5.0,5.0), (6.0,6.0))
Now, our (simple) linear regression model will be parameterised by an intercept, alpha, a slope, beta, and a residual variance, v. So, for convenience, let’s define an ADT representing a particular linear model.
case class Param(alpha: Double, beta: Double, v: Double)
// defined class Param
Now we can define a prior distribution over models as follows.
val prior = for {
alpha <- Normal(0,10)
beta <- Normal(0,4)
v <- Gamma(1,0.1)
} yield Param(alpha, beta, v)
// prior: Wrapped.Prob[Param] = Empirical(Vector(Particle(Param(-2.392517550699654,-3.7516090283880095,1.724680963054379),0.0), Particle(Param(7.60982717067903,-1.4318199629361292,2.9436745225038545),0.0), Particle(Param(-1.0281832158124837,-0.2799562317845073,4.05125312048092),0.0), Particle(Param(-1.0509321093485073,-2.4733837587060448,0.5856868459456287),0.0), Particle(Param(7.678898742733517,0.15616204936412104,5.064540017623097),0.0), Particle(Param(-3.392028985658713,-0.694412176170572,7.452625596437611),0.0), Particle(Param(3.0310535934425324,-2.97938526497514,2.138446100857938),0.0), Particle(Param(3.016959696424399,1.3370878561954143,6.18957854813488),0.0), Particle(Param(2.6956505371497066,1.058845844793446,5.257973123790336),0.0), Particle(Param(1.496225540527873,-1.573936445746...
Since our language doesn’t include any direct syntactic support for fitting regression models, we can define our own function for conditioning a distribution over models on a data point, which we can then apply to our prior as a fold over the available data.
def addPoint(current: Prob[Param], obs: (Double, Double)): Prob[Param] = for {
p <- current
(x, y) = obs
_ <- Normal(p.alpha + p.beta * x, p.v).fitQ(y)
} yield p
// addPoint: (current: Wrapped.Prob[Param], obs: (Double, Double))Wrapped.Prob[Param]
val mod = xy.foldLeft(prior)(addPoint(_,_)).empirical
// mod: Vector[Param] = Vector(Param(1.4386051853067798,0.8900831186754122,4.185564696221981), Param(0.5530582357040271,1.1296886766045509,3.468527573093037), Param(0.6302560079049571,0.9396563485293532,3.7044543917875927), Param(3.68291303096638,0.4781372802435529,5.151665328789926), Param(3.016959696424399,0.4438016738989412,1.9988914122633519), Param(3.016959696424399,0.4438016738989412,1.9988914122633519), Param(0.6302560079049571,0.9396563485293532,3.7044543917875927), Param(0.6302560079049571,0.9396563485293532,3.7044543917875927), Param(3.68291303096638,0.4781372802435529,5.151665328789926), Param(3.016959696424399,0.4438016738989412,1.9988914122633519), Param(0.6302560079049571,0.9396563485293532,3.7044543917875927), Param(0.6302560079049571,0.9396563485293532,3.7044543917875927), ...
meanVar(mod map (_.alpha))
// res4: breeze.stats.meanAndVariance.MeanAndVariance = MeanAndVariance(1.5740812481283812,1.893684802867127,300)
meanVar(mod map (_.beta))
// res5: breeze.stats.meanAndVariance.MeanAndVariance = MeanAndVariance(0.7690238868623273,0.1054479268115053,300)
meanVar(mod map (_.v))
// res6: breeze.stats.meanAndVariance.MeanAndVariance = MeanAndVariance(3.5240853748668695,2.793386340338213,300)
We could easily add syntactic support to our language to enable the fitting of regression-style models, as is done in Rainier, of which more later.
Dynamic generalised linear model
The previous examples have been fairly simple, so let’s finish with something a bit less trivial. Our language is quite flexible enough to allow the analysis of a dynamic generalised linear model (DGLM). Here we’ll fit a Poisson DGLM with a log-link and a simple Brownian state evolution. More complex models are more-or-less similarly straightforward. The model is parameterised by an initial state, state0, and and evolution variance, w.
val data = List(2,1,0,2,3,4,5,4,3,2,1)
// data: List[Int] = List(2, 1, 0, 2, 3, 4, 5, 4, 3, 2, 1)
val prior = for {
w <- Gamma(1, 1)
state0 <- Normal(0.0, 2.0)
} yield (w, List(state0))
// prior: Wrapped.Prob[(Double, List[Double])] = Empirical(Vector(Particle((0.12864918092587044,List(-2.862479260552014)),0.0), Particle((1.1706344622093179,List(1.6138397233532091)),0.0), Particle((0.757288087950638,List(-0.3683499919402798)),0.0), Particle((2.755201217523856,List(-0.6527488751780317)),0.0), Particle((0.7535085397802043,List(0.5135562407906502)),0.0), Particle((1.1630726564525629,List(0.9703146201262348)),0.0), Particle((1.0080345715326213,List(-0.375686732266234)),0.0), Particle((4.603723117526974,List(-1.6977366375222938)),0.0), Particle((0.2870669117815037,List(2.2732160435099433)),0.0), Particle((2.454675218313211,List(-0.4148287542786906)),0.0), Particle((0.3612534201761152,List(-1.0099270904161748)),0.0), Particle((0.29578453393473114,List(-2.4938128878051966)),0.0)...
We can define a function to create a new hidden state, prepend it to the list of hidden states, and condition on the observed value at that time point as follows.
def addTimePoint(current: Prob[(Double, List[Double])],
obs: Int): Prob[(Double, List[Double])] = for {
tup <- current
(w, states) = tup
os = states.head
ns <- Normal(os, w)
_ <- Poisson(math.exp(ns)).fitQ(obs)
} yield (w, ns :: states)
// addTimePoint: (current: Wrapped.Prob[(Double, List[Double])], obs: Int)Wrapped.Prob[(Double, List[Double])]
We then run our (augmented state) particle filter as a fold over the time series.
val mod = data.foldLeft(prior)(addTimePoint(_,_)).empirical
// mod: Vector[(Double, List[Double])] = Vector((0.053073252551193446,List(0.8693030057529023, 1.2746526177834938, 1.020307245610461, 1.106341696651584, 1.070777529635013, 0.8749041525303247, 0.9866999164354662, 0.4082577920509255, 0.06903234462140699, -0.018835642776197814, -0.16841912034400547, -0.08919045681401294)), (0.0988871875952762,List(-0.24241948109998607, 0.09321618969352086, 0.9650532206325375, 1.1738734442767293, 1.2272325310228442, 0.9791695328246326, 0.5576319082578128, -0.0054280215024367084, 0.4256621012454391, 0.7486862644576158, 0.8193517409118243, 0.5928750312493785)), (0.16128799384962295,List(-0.30371187329667104, -0.3976854602292066, 0.5869357473774455, 0.9881090696832543, 1.2095181380307558, 0.7211231597865506, 0.8085486452269925, 0.2664373341459165, -0.627344024142...
meanVar(mod map (_._1)) // w
// res7: breeze.stats.meanAndVariance.MeanAndVariance = MeanAndVariance(0.29497487517435844,0.0831412016262515,300)
meanVar(mod map (_._2.reverse.head)) // state0 (initial state)
// res8: breeze.stats.meanAndVariance.MeanAndVariance = MeanAndVariance(0.04617218427664018,0.372844704533101,300)
meanVar(mod map (_._2.head)) // stateN (final state)
// res9: breeze.stats.meanAndVariance.MeanAndVariance = MeanAndVariance(0.4937178761565612,0.2889287607470016,300)
Summary, conclusions, and further reading
So, we’ve seen how we can build a fully functional, general-purpose, compositional, monadic PPL from scratch in 50 lines of code, and we’ve seen how we can use it to solve real, analytically intractable Bayesian inference problems of non-trivial complexity. Of course, there are many limitations to using exactly this PPL implementation in practice. The algorithm becomes intolerably slow for deeply nested models, and uses unreasonably large amounts of RAM for large numbers of particles. It also suffers from a particle degeneracy problem if there are too many conditioning events. But it is important to understand that these are all deficiencies of the naive inference algorithm used, not the PPL itself. The PPL is flexible and compositional and can be used to build models of arbitrary size and complexity – it just needs to be underpinned by a better, more efficient, inference algorithm. Rainier is a Scala library I’ve blogged about previously which uses a very similar PPL to the one described here, but is instead underpinned by a fast, efficient, HMC algorithm. With my student Jonny Law, we have recently arXived a paper on Functional probabilistic programming for scalable Bayesian modelling, discussing some of these issues, and exploring the compositional nature of monadic PPLs (somewhat glossed over in this post).
Since the same PPL can be underpinned by different inference algorithms encapsulated as probability monads, an obvious question is whether it is possible to abstract the PPL away from the inference algorithm implementation. Of course, the answer is “yes”, and this has been explored to great effect in papers such as Practical probabilistic programming with monads and Functional programming for modular Bayesian inference. Note that they use the cond approach to conditioning, which looks a bit unwieldy, but is equivalent to fitting. As well as allowing alternative inference algorithms to be applied to the same probabilistic program, it also enables the composing of inference algorithms – for example, composing a MH algorithm with an SMC algorithm in order to get a PMMH algorithm. The ideas are implemented in an embedded DSL for Haskell, monad-bayes. If you are not used to Haskell, the syntax will probably seem a bit more intimidating than Scala’s, but the semantics are actually quite similar, with the main semantic difference being that Scala is strictly evaluated by default, whereas Haskell is lazily evaluated by default. Both languages support both lazy and strict evaluation – the difference relates simply to default behaviour, but is important nevertheless.
Papers
Software
- min-ppl – code associated with this blog post
- Rainier – a more efficient PPL with similar syntax
- monad-bayes – a Haskell library exploring related ideas

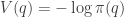
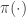


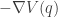

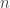

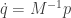
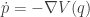

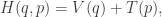
![\displaystyle \dot{H} = \nabla V\cdot \dot{q} + \dot{p}^\text{T}M^{-1}p = \nabla V\cdot \dot{q} + \dot{p}^\text{T}\dot{q} = [\nabla V + \dot{p}]\cdot\dot{q} = 0.](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cdot%7BH%7D+%3D+%5Cnabla+V%5Ccdot+%5Cdot%7Bq%7D+%2B+%5Cdot%7Bp%7D%5E%5Ctext%7BT%7DM%5E%7B-1%7Dp+%3D+%5Cnabla+V%5Ccdot+%5Cdot%7Bq%7D+%2B+%5Cdot%7Bp%7D%5E%5Ctext%7BT%7D%5Cdot%7Bq%7D+%3D+%5B%5Cnabla+V+%2B+%5Cdot%7Bp%7D%5D%5Ccdot%5Cdot%7Bq%7D+%3D+0.+&bg=ffffff&fg=333333&s=0&c=20201002)
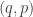
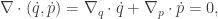
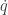
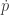
![\displaystyle \tilde \pi(q,p) \propto \exp[-H(q,p)]](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Ctilde+%5Cpi%28q%2Cp%29+%5Cpropto+%5Cexp%5B-H%28q%2Cp%29%5D+&bg=ffffff&fg=333333&s=0&c=20201002)
![\displaystyle \tilde \pi(q,p) \propto \exp[-V(q)]\exp[-T(p)],](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Ctilde+%5Cpi%28q%2Cp%29+%5Cpropto+%5Cexp%5B-V%28q%29%5D%5Cexp%5B-T%28p%29%5D%2C+&bg=ffffff&fg=333333&s=0&c=20201002)
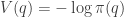
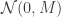




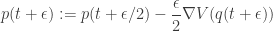

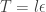
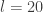
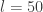
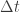 . We can improve the accuracy of this approximation by making the step size smaller, but this will come at the expense of a more slowly mixing MCMC chain.
. We can improve the accuracy of this approximation by making the step size smaller, but this will come at the expense of a more slowly mixing MCMC chain.![\displaystyle l(\beta;y) = y^\textsf{T}X\beta - \mathbf{1}^\textsf{T}\log(\mathbf{1}+\exp[X\beta])](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+l%28%5Cbeta%3By%29+%3D+y%5E%5Ctextsf%7BT%7DX%5Cbeta+-+%5Cmathbf%7B1%7D%5E%5Ctextsf%7BT%7D%5Clog%28%5Cmathbf%7B1%7D%2B%5Cexp%5BX%5Cbeta%5D%29+&bg=ffffff&fg=333333&s=0&c=20201002)
![\displaystyle l(\beta;y) = \sum_j\sum_j y_iX_{ij}\beta_j - \sum_i\log\left(1+\exp\left[\sum_jX_{ij}\beta_j\right]\right).](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+l%28%5Cbeta%3By%29+%3D+%5Csum_j%5Csum_j+y_iX_%7Bij%7D%5Cbeta_j+-+%5Csum_i%5Clog%5Cleft%281%2B%5Cexp%5Cleft%5B%5Csum_jX_%7Bij%7D%5Cbeta_j%5Cright%5D%5Cright%29.+&bg=ffffff&fg=333333&s=0&c=20201002)
 gives
gives![\displaystyle \frac{\partial l}{\partial \beta_k} = \sum_i y_iX_{ik} - \sum_i \frac{\exp\left[\sum_j X_{ij}\beta_j\right]X_{ik}}{1+\exp\left[\sum_j X_{ij}\beta_j\right]}.](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cfrac%7B%5Cpartial+l%7D%7B%5Cpartial+%5Cbeta_k%7D+%3D+%5Csum_i+y_iX_%7Bik%7D+-+%5Csum_i+%5Cfrac%7B%5Cexp%5Cleft%5B%5Csum_j+X_%7Bij%7D%5Cbeta_j%5Cright%5DX_%7Bik%7D%7D%7B1%2B%5Cexp%5Cleft%5B%5Csum_j+X_%7Bij%7D%5Cbeta_j%5Cright%5D%7D.+&bg=ffffff&fg=333333&s=0&c=20201002)
![\displaystyle \nabla l = X^\textsf{T}\left[ y - \frac{\mathbf{1}}{\mathbf{1}+\exp[-X\beta]} \right].](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cnabla+l+%3D+X%5E%5Ctextsf%7BT%7D%5Cleft%5B+y+-+%5Cfrac%7B%5Cmathbf%7B1%7D%7D%7B%5Cmathbf%7B1%7D%2B%5Cexp%5B-X%5Cbeta%5D%7D+%5Cright%5D.+&bg=ffffff&fg=333333&s=0&c=20201002)
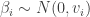 priors, it is easy to see that the gradient of the log prior is just
priors, it is easy to see that the gradient of the log prior is just  . It is the sum of these two terms that gives the gradient of the log posterior.
. It is the sum of these two terms that gives the gradient of the log posterior.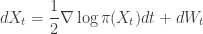

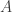 , the SDE
, the SDE
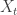 on a common scale.
on a common scale.
 is said to satisfy
is said to satisfy  if
if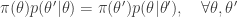
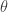 gives
gives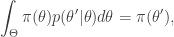
 that satisfies detailed balance. This will then give us a way to (asymptotically) generate samples from our target.
that satisfies detailed balance. This will then give us a way to (asymptotically) generate samples from our target. . Here we drop the notational dependence on
. Here we drop the notational dependence on 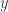 , since MCMC can be used for any target distribution of interest.
, since MCMC can be used for any target distribution of interest. and a target of interest,
and a target of interest,  to construct a new transition kernel
to construct a new transition kernel  can be described algorithmically as follows:
can be described algorithmically as follows: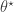 by simulating from
by simulating from  .
.![\displaystyle \alpha(\theta^\star|\theta) = \min\left[1,\frac{\pi(\theta^\star)q(\theta|\theta^\star)}{\pi(\theta)q(\theta^\star|\theta)}\right].](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Calpha%28%5Ctheta%5E%5Cstar%7C%5Ctheta%29+%3D+%5Cmin%5Cleft%5B1%2C%5Cfrac%7B%5Cpi%28%5Ctheta%5E%5Cstar%29q%28%5Ctheta%7C%5Ctheta%5E%5Cstar%29%7D%7B%5Cpi%28%5Ctheta%29q%28%5Ctheta%5E%5Cstar%7C%5Ctheta%29%7D%5Cright%5D.+&bg=ffffff&fg=333333&s=0&c=20201002)
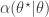 return new state
return new state  , otherwise return
, otherwise return 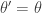 .
.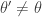 the transition kernel will be
the transition kernel will be 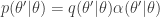 . But then
. But then![\displaystyle \pi(\theta)p(\theta'|\theta) = \min[\pi(\theta)q(\theta'|\theta),\pi(\theta')q(\theta|\theta')]](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cpi%28%5Ctheta%29p%28%5Ctheta%27%7C%5Ctheta%29+%3D+%5Cmin%5B%5Cpi%28%5Ctheta%29q%28%5Ctheta%27%7C%5Ctheta%29%2C%5Cpi%28%5Ctheta%27%29q%28%5Ctheta%7C%5Ctheta%27%29%5D+&bg=ffffff&fg=333333&s=0&c=20201002)
 , and so detailed balance is satisfied. Since detailed balance is trivial at the point mass at
, and so detailed balance is satisfied. Since detailed balance is trivial at the point mass at 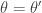 we are done.
we are done. , and so
, and so 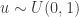 and accept if
and accept if 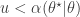 . This is convenient for several reasons. First, it means that we can ignore the "min", and just accept if
. This is convenient for several reasons. First, it means that we can ignore the "min", and just accept if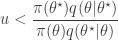
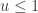 regardless. Better still, we can take logs, and accept if
regardless. Better still, we can take logs, and accept if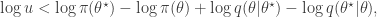
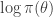 term (which in the context of Bayesian inference will typically be the log posterior) will have been computed at the previous update (irrespective of whether or not the previous move was accepted). So if we are careful about how we pass on that old value to the next iteration, we can avoid recomputing the log posterior, and our algorithm will only require one log posterior evaluation per iteration rather than two. In functional programming languages it is often convenient to pass around this current log posterior density evaluation explicitly, effectively augmenting the state space of the Markov chain to include the log posterior density.
term (which in the context of Bayesian inference will typically be the log posterior) will have been computed at the previous update (irrespective of whether or not the previous move was accepted). So if we are careful about how we pass on that old value to the next iteration, we can avoid recomputing the log posterior, and our algorithm will only require one log posterior evaluation per iteration rather than two. In functional programming languages it is often convenient to pass around this current log posterior density evaluation explicitly, effectively augmenting the state space of the Markov chain to include the log posterior density.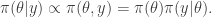
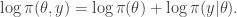
 . We assumed independent mean zero normal priors for the components of this vector, so the log prior is just the sum of logs of normal densities. Many scientific libraries will have built-in functions for returning the log-pdf of standard distributions, but if an explicit form is required, the log of the density of a
. We assumed independent mean zero normal priors for the components of this vector, so the log prior is just the sum of logs of normal densities. Many scientific libraries will have built-in functions for returning the log-pdf of standard distributions, but if an explicit form is required, the log of the density of a 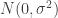 at
at 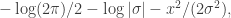
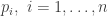 , the likelihood associated with observation
, the likelihood associated with observation  is
is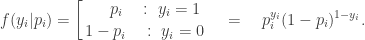
 we have
we have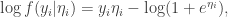

 is the linear predictor, so
is the linear predictor, so  , giving
, giving![\displaystyle \ell(\beta;y) = y^\textsf{T}X\beta - \mathbf{1}^\textsf{T}\log(\mathbf{1}+\exp[X\beta]).](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cell%28%5Cbeta%3By%29+%3D+y%5E%5Ctextsf%7BT%7DX%5Cbeta+-+%5Cmathbf%7B1%7D%5E%5Ctextsf%7BT%7D%5Clog%28%5Cmathbf%7B1%7D%2B%5Cexp%5BX%5Cbeta%5D%29.+&bg=ffffff&fg=333333&s=0&c=20201002)
 is 1 for success and -1 for failure. This isn’t really a problem, since the two encodings are related by
is 1 for success and -1 for failure. This isn’t really a problem, since the two encodings are related by  . This new encoding is convenient in the context of a logit parameterisation since then
. This new encoding is convenient in the context of a logit parameterisation since then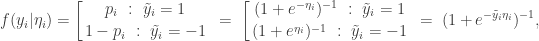

![\displaystyle \ell(\eta;\tilde y) = -\mathbf{1}\cdot \log(\mathbf{1}+\exp[-\tilde y\circ \eta]),](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cell%28%5Ceta%3B%5Ctilde+y%29+%3D+-%5Cmathbf%7B1%7D%5Ccdot+%5Clog%28%5Cmathbf%7B1%7D%2B%5Cexp%5B-%5Ctilde+y%5Ccirc+%5Ceta%5D%29%2C+&bg=ffffff&fg=333333&s=0&c=20201002)
 denotes the
denotes the ![\displaystyle \ell(\beta;\tilde y) = -\mathbf{1}^\textsf{T} \log(\mathbf{1}+\exp[-\tilde y\circ X\beta]).](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cell%28%5Cbeta%3B%5Ctilde+y%29+%3D+-%5Cmathbf%7B1%7D%5E%5Ctextsf%7BT%7D+%5Clog%28%5Cmathbf%7B1%7D%2B%5Cexp%5B-%5Ctilde+y%5Ccirc+X%5Cbeta%5D%29.+&bg=ffffff&fg=333333&s=0&c=20201002)
![\displaystyle \ell(\beta; y) = -\mathbf{1}^\textsf{T} \log(\mathbf{1}+\exp[-(2y-\mathbf{1})\circ (X\beta)]),](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cell%28%5Cbeta%3B+y%29+%3D+-%5Cmathbf%7B1%7D%5E%5Ctextsf%7BT%7D+%5Clog%28%5Cmathbf%7B1%7D%2B%5Cexp%5B-%282y-%5Cmathbf%7B1%7D%29%5Ccirc+%28X%5Cbeta%29%5D%29%2C+&bg=ffffff&fg=333333&s=0&c=20201002)
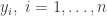 are 1 for a "success" and 0 for a "failure". The covariate
are 1 for a "success" and 0 for a "failure". The covariate 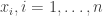 all have 1 as the first element. The statistical model is
all have 1 as the first element. The statistical model is![\displaystyle \text{logit}\left(\text{Pr}[Y_i = 1]\right) = x_i \cdot \beta,\quad i=1,\ldots,n,](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Ctext%7Blogit%7D%5Cleft%28%5Ctext%7BPr%7D%5BY_i+%3D+1%5D%5Cright%29+%3D+x_i+%5Ccdot+%5Cbeta%2C%5Cquad+i%3D1%2C%5Cldots%2Cn%2C+&bg=ffffff&fg=333333&s=0&c=20201002)
 , and
, and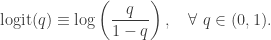
![\displaystyle \text{Pr}[Y_i = 1] = \text{expit}(x_i \cdot \beta),\quad i=1,\ldots,n,](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Ctext%7BPr%7D%5BY_i+%3D+1%5D+%3D+%5Ctext%7Bexpit%7D%28x_i+%5Ccdot+%5Cbeta%29%2C%5Cquad+i%3D1%2C%5Cldots%2Cn%2C+&bg=ffffff&fg=333333&s=0&c=20201002)
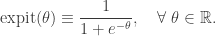
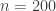 observations and 7 predictors. Adding an intercept gives
observations and 7 predictors. Adding an intercept gives  covariates. For the Bayesian analysis, we need a prior on
covariates. For the Bayesian analysis, we need a prior on 










