Part 1: The basics
Introduction
This is the first in a series of posts on MCMC-based fully Bayesian inference for a logistic regression model. In this series we will look at the model, and see how the posterior distribution can be sampled using a variety of different programming languages and libraries.
Logistic regression
Logistic regression is concerned with predicting a binary outcome based on some covariate information. The probability of "success" is modelled via a logistic transformation of a linear predictor constructed from the covariate vector.
This is a very simple model, but is a convenient toy example since it is arguably the simplest interesting example of an intractable (nonlinear) statistical model requiring some kind of iterative numerical fitting method, even in the non-Bayesian setting. In a Bayesian context, the posterior distribution is intractable, necessitating either approximate or computationally intensive numerical methods of "solution". In this series of posts, we will mainly concentrate on MCMC algortithms for sampling the full posterior distribution of the model parameters given some observed data.
We assume 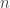

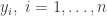
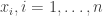
![\displaystyle \text{logit}\left(\text{Pr}[Y_i = 1]\right) = x_i \cdot \beta,\quad i=1,\ldots,n,](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Ctext%7Blogit%7D%5Cleft%28%5Ctext%7BPr%7D%5BY_i+%3D+1%5D%5Cright%29+%3D+x_i+%5Ccdot+%5Cbeta%2C%5Cquad+i%3D1%2C%5Cldots%2Cn%2C+&bg=ffffff&fg=333333&s=0&c=20201002)
where 

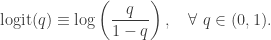
Equivalently,
![\displaystyle \text{Pr}[Y_i = 1] = \text{expit}(x_i \cdot \beta),\quad i=1,\ldots,n,](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Ctext%7BPr%7D%5BY_i+%3D+1%5D+%3D+%5Ctext%7Bexpit%7D%28x_i+%5Ccdot+%5Cbeta%29%2C%5Cquad+i%3D1%2C%5Cldots%2Cn%2C+&bg=ffffff&fg=333333&s=0&c=20201002)
where
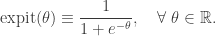
Note that the expit function is sometimes called the logistic or sigmoid function, but expit is slightly less ambiguous. The statistical problem is to choose the parameter vector
Example problem
In order to illustrate the ideas, it is useful to have a small running example. Here we will use the (infamous) Pima training dataset (MASS::Pima.tr in R). Here there are 

Describing the model in some PPLs
In this first post in the series, we will use probabilistic programming languages (PPLs) to describe the model and sample the posterior distribution.
JAGS
JAGS is a stand-alone domain specific language (DSL) for probabilistic programming. It can be used independently of general purpose programming languages, or called from popular languages for data science such as Python and R. We can describe our model in JAGS with the following code.
model {
for (i in 1:n) {
y[i] ~ dbern(pr[i])
logit(pr[i]) <- inprod(X[i,], beta)
}
beta[1] ~ dnorm(0, 0.01)
for (j in 2:p) {
beta[j] ~ dnorm(0, 1)
}
}
Note that JAGS uses precision as the second parameter of a normal distribution. See the full runnable R script for further details. Given this model description, JAGS can construct an MCMC sampler for the posterior distribution of the model parameters given the data. See the full script for how to feed in the data, run the sampler, and analyse the output.
Stan
Stan is another stand-alone DSL for probabilistic programming, and has a very sophisticated sampling algorithm, making it a popular choice for non-trivial models. It uses gradient information for sampling, and therefore requires a differentiable log-posterior. We could encode our logistic regression model as follows.
data {
int<lower=1> n;
int<lower=1> p;
int<lower=0, upper=1> y[n];
real X[n,p];
}
parameters {
real beta[p];
}
model {
for (i in 1:n) {
real eta = dot_product(beta, X[i,]);
real pr = 1/(1+exp(-eta));
y[i] ~ binomial(1, pr);
}
beta[1] ~ normal(0, 10);
for (j in 2:p) {
beta[j] ~ normal(0, 1);
}
}
Note that Stan uses standard deviation as the second parameter of the normal distribution. See the full runnable R script for further details.
PyMC
JAGS and Stan are stand-alone DSLs for probabilistic programming. This has the advantage of making them independent of any particular host (general purpose) programming language. But it also means that they are not able to take advantage of the language and tool support of an existing programming language. An alternative to stand-alone DSLs are embedded DSLs (eDSLs). Here, a DSL is embedded as a library or package within an existing (general purpose) programming language. Then, ideally, in the context of PPLs, probabilistic programs can become ordinary values within the host language, and this can have some advantages, especially if the host language is sophisticated. A number of probabilistic programming languages have been implemented as eDSLs in Python. Python is not a particularly sophisticated language, so the advantages here are limited, but not negligible.
PyMC is probably the most popular eDSL PPL in Python. We can encode our model in PyMC as follows.
pscale = np.array([10.,1.,1.,1.,1.,1.,1.,1.])
with pm.Model() as model:
beta = pm.Normal('beta', 0, pscale, shape=p)
eta = pmm.matrix_dot(X, beta)
pm.Bernoulli('y', logit_p=eta, observed=y)
traces = pm.sample(2500, tune=1000, init="adapt_diag", return_inferencedata=True)
See the full runnable script for further details.
NumPyro
NumPyro is a fork of Pyro for NumPy and JAX (of which more later). We can encode our model with NumPyro as follows.
pscale = jnp.array([10.,1.,1.,1.,1.,1.,1.,1.]).astype(jnp.float32)
def model(X, y):
coefs = numpyro.sample("beta", dist.Normal(jnp.zeros(p), pscale))
logits = jnp.dot(X, coefs)
return numpyro.sample("obs", dist.Bernoulli(logits=logits), obs=y)
See the full runnable script for further details.
Please note that none of the above examples have been optimised, or are even necessarily expressed idiomatically within each PPL. I’ve just tried to express the model in as simple and similar way across each PPL. For example, I know that there is a function bernoulli_logit_glm in Stan which would simplify the model and improve sampling efficiency, but I’ve deliberately not used it in order to try and keep the implementations as basic as possible. The same will be true for all of the code examples in this series of blog posts. The code has not been optimised and should therefore not be used for serious benchmarking.
Next steps
PPLs are convenient, and are becoming increasingly sophisticated. Each of the above PPLs provides a simple way to pass in observed data, and automatically construct an MCMC algorithm for sampling from the implied posterior distribution – see the full scripts for details. All of the PPLs work well for this problem, and all produce identical results up to Monte Carlo error. Each PPL has its own approach to sampler construction, and some PPLs offer multiple choices. However, more challenging problems often require highly customised samplers. Such samplers will often need to be created from scratch, and will require (at least) the ability to compute the (unnormalised log) posterior density at proposed parameter values, so in the next post we will look at how this can be derived for this model (in a couple of different ways) and coded up from scratch in a variety of programming languages.
All of the complete, runnable code associated with this series of blog posts can be obtained from this public github repo.
 from the prior and then sampling a corresponding data set
from the prior and then sampling a corresponding data set 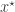 from the model. This simulated data set is compared with the true data
from the model. This simulated data set is compared with the true data  using some (pseudo-)norm,
using some (pseudo-)norm,  , and accepting
, and accepting  . It should be clear that if we are using a proper norm then as
. It should be clear that if we are using a proper norm then as  tends to zero the distribution of the accepted values tends to the desired posterior distribution of the parameters given the data.
tends to zero the distribution of the accepted values tends to the desired posterior distribution of the parameters given the data.
 for
for 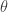 , then it should be clear that replacing the acceptance criterion with
, then it should be clear that replacing the acceptance criterion with  will also lead to a scheme tending to the true posterior as
will also lead to a scheme tending to the true posterior as 
 , and a forwards-simulation model for the data
, and a forwards-simulation model for the data 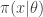 . It is clear that a simple algorithm for simulating from the desired posterior
. It is clear that a simple algorithm for simulating from the desired posterior  can be obtained as follows. First simulate from the joint distribution
can be obtained as follows. First simulate from the joint distribution 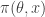 by simulating
by simulating  and then
and then  . This gives a sample
. This gives a sample  from the joint distribution. A simple rejection algorithm which rejects the proposed pair unless
from the joint distribution. A simple rejection algorithm which rejects the proposed pair unless 

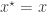 , keep
, keep  , keep
, keep 
 (ideally a sufficient statistic), and accepting provided that
(ideally a sufficient statistic), and accepting provided that 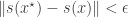 for some suitable choice of metric and
for some suitable choice of metric and  . Note that I have deliberately adopted a functional programming style, making use of the lapply function for the most computationally intensive steps. The reason for this will soon become apparent. Note that rather than pre-specifying a cutoff
. Note that I have deliberately adopted a functional programming style, making use of the lapply function for the most computationally intensive steps. The reason for this will soon become apparent. Note that rather than pre-specifying a cutoff