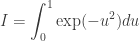Introduction
In the previous post I gave a quick introduction to some important concepts in functional programming, such as HOFs, closures, currying and partial application, and hopefully gave some insight into why these concepts might be useful in the context of scientific computing. Another concept that is very important in modern functional programming is that of the monad. Monads are one of those concepts that turns out to be very simple and intuitive once you “get it”, but completely impenetrable until you do! Now, there zillions of monad tutorials out there, and I don’t think that I have anything particularly insightful to add to the discussion. That said, most of the tutorials focus on problems and examples that are some way removed from the interests of statisticians and scientific programmers. So in this post I want to try and give a very informal and intuitive introduction to the monad concept in a way that I hope will resonate with people from a more scientific computing background.
The term “monad” is borrowed from that of the corresponding concept in category theory. The connection between functional programming and category theory is strong and deep. I intend to expore this more in future posts, but for this post the connection is not important and no knowledge of category theory is assumed (or imparted!).
Functors and Monads
Maps and Functors
All of the code used in this post in contained in the first-monads directory of my blog repo. The best way to follow this post is to copy-and-paste commands one-at-a-time from this post to a Scala REPL or sbt console. Note that only the numerical linear algebra examples later in this post require any non-standard dependencies.
The map method is one of the first concepts one meets when beginning functional programming. It is a higher order method on many (immutable) collection and other container types. Let’s start by looking at how map operates on Lists.
val x = (0 to 4).toList
// x: List[Int] = List(0, 1, 2, 3, 4)
val x2 = x map { x => x * 3 }
// x2: List[Int] = List(0, 3, 6, 9, 12)
val x3 = x map { _ * 3 }
// x3: List[Int] = List(0, 3, 6, 9, 12)
val x4 = x map { _ * 0.1 }
// x4: List[Double] = List(0.0, 0.1, 0.2, 0.30000000000000004, 0.4)
The last example shows that a List[T] can be converted to a List[S] if map is passed a function of type T => S. Of course there’s nothing particularly special about List here. It works with other collection types in the same way, as the following example with (immutable) Vector illustrates:
val xv = x.toVector
// xv: Vector[Int] = Vector(0, 1, 2, 3, 4)
val xv2 = xv map { _ * 0.2 }
// xv2: scala.collection.immutable.Vector[Double] = Vector(0.0, 0.2, 0.4, 0.6000000000000001, 0.8)
val xv3 = for (xi <- xv) yield (xi * 0.2)
// xv3: scala.collection.immutable.Vector[Double] = Vector(0.0, 0.2, 0.4, 0.6000000000000001, 0.8)
Note here that the for comprehension generating xv3 is exactly equivalent to the map call generating xv2 – the for-comprehension is just syntactic sugar for the map call. The benefit of this syntax will become apparent in the more complex examples we consider later.
Many collection and other container types have a map method that behaves this way. Any parametrised type that does have a map method like this is known as a Functor. Again, the name is due to category theory, but that doesn’t matter for this post. From a Scala-programmer perspective, a functor can be thought of as a trait, in pseudo-code as
trait F[T] {
def map(f: T => S): F[S]
}
with F representing the functor. In fact it turns out to be better to think of a functor as a type class, but that is yet another topic for a future post… Also note that to be a functor in the strict sense (from a category theory perspective), the map method must behave sensibly – that is, it must satisfy the functor laws. But again, I’m keeping things informal and intuitive for this post – there are plenty of other monad tutorials which emphasise the category theory connections.
FlatMap and Monads
Once we can map functions over elements of containers, we soon start mapping functions which themselves return values of the container type. eg. we can map a function returning a List over the elements of a List, as illustrated below.
val x5 = x map { x => List(x - 0.1, x + 0.1) }
// x5: List[List[Double]] = List(List(-0.1, 0.1), List(0.9, 1.1), List(1.9, 2.1), List(2.9, 3.1), List(3.9, 4.1))
Clearly this returns a list-of-lists. Sometimes this is what we want, but very often we actually want to flatten down to a single list so that, for example, we can subsequently map over all of the elements of the base type with a single map. We could take the list-of-lists and then flatten it, but this pattern is so common that the act of mapping and then flattening is often considered to be a basic operation, often known in Scala as flatMap. So for our toy example, we could carry out the flatMap as follows.
val x6 = x flatMap { x => List(x - 0.1, x + 0.1) }
// x6: List[Double] = List(-0.1, 0.1, 0.9, 1.1, 1.9, 2.1, 2.9, 3.1, 3.9, 4.1)
The ubiquity of this pattern becomes more apparent when we start thinking about iterating over multiple collections. For example, suppose now that we have two lists, x and y, and that we want to iterate over all pairs of elements consisting of one element from each list.
val y = (0 to 12 by 2).toList
// y: List[Int] = List(0, 2, 4, 6, 8, 10, 12)
val xy = x flatMap { xi => y map { yi => xi * yi } }
// xy: List[Int] = List(0, 0, 0, 0, 0, 0, 0, 0, 2, 4, 6, 8, 10, 12, 0, 4, 8, 12, 16, 20, 24, 0, 6, 12, 18, 24, 30, 36, 0, 8, 16, 24, 32, 40, 48)
This pattern of having one or more nested flatMaps followed by a final map in order to iterate over multiple collections is very common. It is exactly this pattern that the for-comprehension is syntactic sugar for. So we can re-write the above using a for-comprehension
val xy2 = for {
xi <- x
yi <- y
} yield (xi * yi)
// xy2: List[Int] = List(0, 0, 0, 0, 0, 0, 0, 0, 2, 4, 6, 8, 10, 12, 0, 4, 8, 12, 16, 20, 24, 0, 6, 12, 18, 24, 30, 36, 0, 8, 16, 24, 32, 40, 48)
This for-comprehension (usually called a for-expression in Scala) has an intuitive syntax reminiscent of the kind of thing one might write in an imperative language. But it is important to remember that <- is not actually an imperative assignment. The for-comprehension really does expand to the pure-functional nested flatMap and map call given above.
Recalling that a functor is a parameterised type with a map method, we can now say that a monad is just a functor which also has a flatMap method. We can write this in pseudo-code as
trait M[T] {
def map(f: T => S): M[S]
def flatMap(f: T => M[S]): M[S]
}
Not all functors can have a flattening operation, so not all functors are monads, but all monads are functors. Monads are therefore more powerful than functors. Of course, more power is not always good. The principle of least power is one of the main principles of functional programming, but monads are useful for sequencing dependent computations, as illustrated by for-comprehensions. In fact, since for-comprehensions de-sugar to calls to map and flatMap, monads are precisely what are required in order to be usable in for-comprehensions. Collections supporting map and flatMap are referred to as monadic. Most Scala collections are monadic, and operating on them using map and flatMap operations, or using for-comprehensions is referred to as monadic-style. People will often refer to the monadic nature of a collection (or other container) using the word monad, eg. the “List monad”.
So far the functors and monads we have been working with have been collections, but not all monads are collections, and in fact collections are in some ways atypical examples of monads. Many monads are containers or wrappers, so it will be useful to see examples of monads which are not collections.
Option monad
One of the first monads that many people encounter is the Option monad (referred to as the Maybe monad in Haskell, and Optional in Java 8). You can think of it as being a strange kind of “collection” that can contain at most one element. So it will either contain an element or it won’t, and so can be used to wrap the result of a computation which might fail. If the computation succeeds, the value computed can be wrapped in the Option (using the type Some), and if it fails, it will not contain a value of the required type, but simply be the value None. It provides a referentially transparent and type-safe alternative to raising exceptions or returning NULL references. We can transform Options using map.
val three = Option(3)
// three: Option[Int] = Some(3)
val twelve = three map (_ * 4)
// twelve: Option[Int] = Some(12)
But when we start combining the results of multiple computations that could fail, we run into exactly the same issues as before.
val four = Option(4)
// four: Option[Int] = Some(4)
val twelveB = three map (i => four map (i * _))
// twelveB: Option[Option[Int]] = Some(Some(12))
Here we have ended up with an Option wrapped in another Option, which is not what we want. But we now know the solution, which is to replace the first map with flatMap, or better still, use a for-comprehension.
val twelveC = three flatMap (i => four map (i * _))
// twelveC: Option[Int] = Some(12)
val twelveD = for {
i <- three
j <- four
} yield (i * j)
// twelveD: Option[Int] = Some(12)
Again, the for-comprehension is a little bit easier to understand than the chaining of calls to flatMap and map. Note that in the for-comprehension we don’t worry about whether or not the Options actually contain values – we just concentrate on the “happy path”, where they both do, safe in the knowledge that the Option monad will take care of the failure cases for us. Two of the possible failure cases are illustrated below.
val oops: Option[Int] = None
// oops: Option[Int] = None
val oopsB = for {
i <- three
j <- oops
} yield (i * j)
// oopsB: Option[Int] = None
val oopsC = for {
i <- oops
j <- four
} yield (i * j)
// oopsC: Option[Int] = None
This is a typical benefit of code written in a monadic style. We chain together multiple computations thinking only about the canonical case and trusting the monad to take care of any additional computational context for us.
IEEE floating point and NaN
Those with a background in scientific computing are probably already familiar with the NaN value in IEEE floating point. In many regards, this value and the rules around its behaviour mean that Float and Double types in IEEE compliant languages behave as an Option monad with NaN as the None value. This is simply illustrated below.
val nan = Double.NaN
3.0 * 4.0
// res0: Double = 12.0
3.0 * nan
// res1: Double = NaN
nan * 4.0
// res2: Double = NaN
The NaN value arises naturally when computations fail. eg.
val nanB = 0.0 / 0.0
// nanB: Double = NaN
This Option-like behaviour of Float and Double means that it is quite rare to see examples of Option[Float] or Option[Double] in Scala code. But there are some disadvantages of the IEEE approach, as discussed elsewhere. Also note that this only works for Floats and Doubles, and not for other types, including, say, Int.
val nanC=0/0
// This raises a runtime exception!
Option for matrix computations
Good practical examples of scientific computations which can fail crop up frequently in numerical linear algebra, so it’s useful to see how Option can simplify code in that context. Note that the code in this section requires the Breeze library, so should be run from an sbt console using the sbt build file associated with this post.
In statistical applications, one often needs to compute the Cholesky factorisation of a square symmetric matrix. This operation is built into Breeze as the function cholesky. However the factorisation will fail if the matrix provided is not positive semi-definite, and in this case the cholesky function will throw a runtime exception. We will use the built in cholesky function to create our own function, safeChol (using a monad called Try which is closely related to the Option monad) returning an Option of a matrix rather than a matrix. This function will not throw an exception, but instead return None in the case of failure, as illustrated below.
import breeze.linalg._
def safeChol(m: DenseMatrix[Double]): Option[DenseMatrix[Double]] = scala.util.Try(cholesky(m)).toOption
val m = DenseMatrix((2.0, 1.0), (1.0, 3.0))
val c = safeChol(m)
// c: Option[breeze.linalg.DenseMatrix[Double]] =
// Some(1.4142135623730951 0.0
// 0.7071067811865475 1.5811388300841898 )
val m2 = DenseMatrix((1.0, 2.0), (2.0, 3.0))
val c2 = safeChol(m2)
// c2: Option[breeze.linalg.DenseMatrix[Double]] = None
A Cholesky factorisation is often followed by a forward or backward solve. This operation may also fail, independently of whether the Cholesky factorisation fails. There doesn’t seem to be a forward solve function directly exposed in the Breeze API, but we can easily define one, which I call dangerousForwardSolve, as it will throw an exception if it fails, just like the cholesky function. But just as for the cholesky function, we can wrap up the dangerous function into a safe one that returns an Option.
import com.github.fommil.netlib.BLAS.{getInstance => blas}
def dangerousForwardSolve(A: DenseMatrix[Double], y: DenseVector[Double]): DenseVector[Double] = {
val yc = y.copy
blas.dtrsv("L", "N", "N", A.cols, A.toArray, A.rows, yc.data, 1)
yc
}
def safeForwardSolve(A: DenseMatrix[Double], y: DenseVector[Double]): Option[DenseVector[Double]] = scala.util.Try(dangerousForwardSolve(A, y)).toOption
Now we can write a very simple function which chains these two operations together, as follows.
def safeStd(A: DenseMatrix[Double], y: DenseVector[Double]): Option[DenseVector[Double]] = for {
L <- safeChol(A)
z <- safeForwardSolve(L, y)
} yield z
safeStd(m,DenseVector(1.0,2.0))
// res15: Option[breeze.linalg.DenseVector[Double]] = Some(DenseVector(0.7071067811865475, 0.9486832980505138))
Note how clean and simple this function is, concentrating purely on the “happy path” where both operations succeed and letting the Option monad worry about the three different cases where at least one of the operations fails.
The Future monad
Let’s finish with a monad for parallel and asynchronous computation, the Future monad. The Future monad is used for wrapping up slow computations and dispatching them to another thread for completion. The call to Future returns immediately, allowing the calling thread to continue while the additional thread processes the slow work. So at that stage, the Future will not have completed, and will not contain a value, but at some (unpredictable) time in the future it (hopefully) will (hence the name). In the following code snippet I construct two Futures that will each take at least 10 seconds to complete. On the main thread I then use a for-comprehension to chain the two computations together. Again, this will return immediately returning another Future that at some point in the future will contain the result of the derived computation. Then, purely for illustration, I force the main thread to stop and wait for that third future (f3) to complete, printing the result to the console.
import scala.concurrent.duration._
import scala.concurrent.{Future,ExecutionContext,Await}
import ExecutionContext.Implicits.global
val f1=Future{
Thread.sleep(10000)
1 }
val f2=Future{
Thread.sleep(10000)
2 }
val f3=for {
v1 <- f1
v2 <- f2
} yield (v1+v2)
println(Await.result(f3,30.second))
When you paste this into your console you should observe that you get the result in 10 seconds, as f1 and f2 execute in parallel on separate threads. So the Future monad is one (of many) ways to get started with parallel and async programming in Scala.
Summary
In this post I’ve tried to give a quick informal introduction to the monad concept, and tried to use examples that will make sense to those interested in scientific and statistical computing. There’s loads more to say about monads, and there are many more commonly encountered useful monads that haven’t been covered in this post. I’ve skipped over lots of details, especially those relating to the formal definitions of functors and monads, including the laws that map and flatMap must satisfy and why. But those kinds of details can be easily picked up from other monad tutorials. Anyone interested in pursuing the formal connections may be interested in a page of links I’m collating on category theory for FP. In particular, I quite like the series of blog posts on category theory for programmers. As I’ve mentioned in previous posts, I also really like the book Functional Programming in Scala, which I strongly recommend to anyone who wants to improve their Scala code. In a subsequent post I’ll explain how monadic style is relevant to issues relating to the statistical analysis of big data, as exemplified in Apache Spark. It’s probably also worth mentioning that there is another kind of functor that turns out to be exceptionally useful in functional programming: the applicative functor. This is more powerful than a basic functor, but less powerful than a monad. It turns out to be useful for computations which need to be sequenced but are not sequentially dependent (context-free rather than context-sensitive), and is a little bit more general and flexible than a monad in cases where it is appropriate.
 of the process is being updated. That is, we target
of the process is being updated. That is, we target 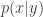 (for known, fixed,
(for known, fixed, 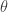 ) rather than
) rather than 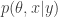 . This special case is known as the particle independent Metropolis-Hastings (PIMH) sampler.
. This special case is known as the particle independent Metropolis-Hastings (PIMH) sampler. using a bootstrap filter, and then accepting the proposal with probability
using a bootstrap filter, and then accepting the proposal with probability 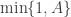 , where
, where 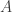 is the
is the 
 is the bootstrap filter’s estimate of marginal likelihood for the new path, and
is the bootstrap filter’s estimate of marginal likelihood for the new path, and  is the estimate associated with the current path. Again using notation from the
is the estimate associated with the current path. Again using notation from the 
 . Again, be sure to see the previous post for the explanation.
. Again, be sure to see the previous post for the explanation.
 rather than the exact conditional distribution
rather than the exact conditional distribution 
![\displaystyle \frac{\tilde{q}(\mathbf{x}_0,\ldots,\mathbf{x}_T,\mathbf{a}_0,\ldots,\mathbf{a}_{T-1})} {\displaystyle p(x_0^{b_0^k})\left[\prod_{t=0}^{T-1} \pi_t^{b_t^k}p\left(x_{t+1}^{b_{t+1}^k}|x_t^{b_t^k}\right)\right]}](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cfrac%7B%5Ctilde%7Bq%7D%28%5Cmathbf%7Bx%7D_0%2C%5Cldots%2C%5Cmathbf%7Bx%7D_T%2C%5Cmathbf%7Ba%7D_0%2C%5Cldots%2C%5Cmathbf%7Ba%7D_%7BT-1%7D%29%7D+%7B%5Cdisplaystyle+p%28x_0%5E%7Bb_0%5Ek%7D%29%5Cleft%5B%5Cprod_%7Bt%3D0%7D%5E%7BT-1%7D+%5Cpi_t%5E%7Bb_t%5Ek%7Dp%5Cleft%28x_%7Bt%2B1%7D%5E%7Bb_%7Bt%2B1%7D%5Ek%7D%7Cx_t%5E%7Bb_t%5Ek%7D%5Cright%29%5Cright%5D%7D&bg=ffffff&fg=333333&s=0&c=20201002) .
.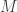 paths, of which the conditioned-on trajectory is one.
paths, of which the conditioned-on trajectory is one.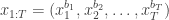 be the path that is to be conditioned on, with ancestral lineage
be the path that is to be conditioned on, with ancestral lineage  . Then, for
. Then, for 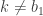 , sample
, sample 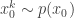 and set
and set  . Now suppose that at time
. Now suppose that at time 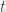 we have a weighted sample from
we have a weighted sample from 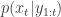 . First resample by sampling
. First resample by sampling 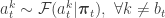 . Next sample
. Next sample  . Then for all
. Then for all 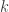 set
set 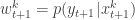 and normalise with
and normalise with 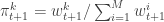 . Propagate this weighted set of particles to the next time point. At time
. Propagate this weighted set of particles to the next time point. At time 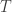 select a single trajectory by sampling
select a single trajectory by sampling  .
. of the
of the  random variables in the augmented state space at each iteration. Since the block of variables to be updated is random, this defines an ergodic sampler for
random variables in the augmented state space at each iteration. Since the block of variables to be updated is random, this defines an ergodic sampler for  particles, and we have explained why the marginal distribution of the selected trajectory is the exact conditional distribution.
particles, and we have explained why the marginal distribution of the selected trajectory is the exact conditional distribution.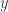 and the currently sampled path,
and the currently sampled path,