Introduction
Very often it is desirable to use Metropolis Hastings MCMC for a target distribution which does not have full support (for example, it may correspond to a non-negative random variable), using a proposal distribution which does (for example, a Gaussian random walk proposal). This isn’t a problem at all, but on more than one occasion now I have come across students getting this wrong, so I thought it might be useful to have a brief post on how to do it right, see what people sometimes get wrong, and why, and then think about correcting the wrong method in order to make it right…
A simple example
For this post we will consider a simple 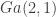

Of course this is a very simple distribution, and there are many straightforward ways to simulate it directly, but for this post we will use a random walk Metropolis-Hastings (MH) scheme with standard Gaussian innovations. So, if the current state of the chain is 
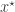
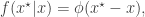
where 
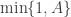

since the standard normal density is symmetric.
Correct implementation
We can easily implement this using R as follows:
met1=function(iters)
{
xvec=numeric(iters)
x=1
for (i in 1:iters) {
xs=x+rnorm(1)
A=dgamma(xs,2,1)/dgamma(x,2,1)
if (runif(1)<A)
x=xs
xvec[i]=x
}
return(xvec)
}
We can run it, plot the results and check it against the true target with the following commands.
iters=1000000 out=met1(iters) hist(out,100,freq=FALSE,main="met1") curve(dgamma(x,2,1),add=TRUE,col=2,lwd=2)

If you have a slow computer, you may prefer to use iters=100000. The above code uses R’s built-in gamma density. Alternatively, we can hard-code the density as follows.
met2=function(iters)
{
xvec=numeric(iters)
x=1
for (i in 1:iters) {
xs=x+rnorm(1)
A=xs*exp(-xs)/(x*exp(-x))
if (runif(1)<A)
x=xs
xvec[i]=x
}
return(xvec)
}
We can run this code using the following commands, to verify that it does work as expected.
out=met2(iters) hist(out,100,freq=FALSE,main="met2") curve(dgamma(x,2,1),add=TRUE,col=2,lwd=2)
However, there is a potential problem with the above code that we have got away with in this instance, which often catches people out. We have hard-coded the density for 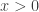
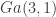
Although this problem often catches people out, it tends not to be a big issue in practice, as it typically leads to an obviously incorrect sampler, or a sampler which crashes, and is relatively simple to debug and fix.
An incorrect sampler
The problem I want to focus on here is more subtle, but closely related. It is clear that any 
met3=function(iters)
{
xvec=numeric(iters)
x=1
for (i in 1:iters) {
repeat {
xs=x+rnorm(1)
if (xs>0)
break
}
A=xs*exp(-xs)/(x*exp(-x))
if (runif(1)<A)
x=xs
xvec[i]=x
}
return(xvec)
}
As reasonable as this idea may at first seem, it does not lead to a sampler having the desired target, as can be verified using the following commands.
out=met3(iters) hist(out,100,freq=FALSE,main="met3") curve(dgamma(x,2,1),add=TRUE,col=2,lwd=2)

So, this sampler seems to be sampling something close to the desired target, but not the same. This raises a couple of questions. First and most important, can we fix this sampler so that it does sample the correct target (yes), and second, can we figure out what target density the incorrect sampler is actually sampling (again, yes)? Let’s start with the issue of how to fix the sampler, as this will also help us to understand what the incorrect sampler is doing.
Fixing the truncated sampler
By repeatedly sampling from the proposal until we obtain a non-negative value, we are actually implementing a rejection sampler for sampling from the proposal distribution truncated at zero. This is a perfectly reasonable proposal distribution, so we can use it provided that we use the correct MH acceptance ratio. Now, the truncated density has the same density as the untruncated density, apart from the differing support and a normalising constant. Indeed, this may be why people often assume this method will work, because normalising constants often don’t matter in MH schemes. However, the normalising constant only doesn’t matter if it is independent of the state, and here it is not… Explicitly, we have

Including the normalising constant we have
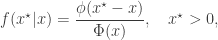
where 

where we see that the normalising constants do not cancel out. We can modify the previous sampler to use the correct acceptance ratio as follows.
met4=function(iters)
{
xvec=numeric(iters)
x=1
for (i in 1:iters) {
repeat {
xs=x+rnorm(1)
if (xs>0)
break
}
A=xs*exp(-xs)/(x*exp(-x))
A=A*pnorm(x)/pnorm(xs)
if (runif(1)<A)
x=xs
xvec[i]=x
}
return(xvec)
}
We can verify that this sampler gives leads to the correct target with the following commands.
out=met4(iters) hist(out,100,freq=FALSE,main="met4") curve(dgamma(x,2,1),add=TRUE,col=2,lwd=2)

So, truncating the proposal at zero is fine, provided that you modify the acceptance ratio accordingly.
What does the incorrect sampler target?
Now that we understand why the naive truncated sampler was wrong and how to fix it, we can, out of curiosity, wonder what distribution that sampler actually targets. Now we understand what proposal we are actually using, we can re-write the acceptance ratio as
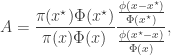
from which it is clear that the actual target of this chain is
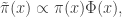
or
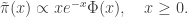
The constant of proportionality is not immediately obvious, but is tractable, and turns out to be a nice undergraduate exercise in integration by parts, leading to

We can verify this using the following commands.
out=met3(iters) hist(out,100,freq=FALSE,main="met3") curve(dgamma(x,2,1)*pnorm(x)*2*sqrt(2*pi)/(sqrt(2*pi)+2),add=TRUE,col=3,lwd=2)

Now we know the actual target of the incorrect sampler, we can compare it with the correct target as follows.
curve(dgamma(x,2,1),0,10,col=2,lwd=2,main="Densities") curve(dgamma(x,2,1)*pnorm(x)*2*sqrt(2*pi)/(sqrt(2*pi)+2),add=TRUE,col=3,lwd=2)

So we see that the distributions are different, but not so different that one would immediate suspect an error on the basis of a sample of output. This makes it a difficult bug to track down.
Summary
There is no problem in principle using a proposal with full support for a target with limited support in MH algorithms. However, it is important to check whether a proposed value is within the support of the target and reject the proposed move if it is not. If you are concerned that such a scheme might be inefficient, it is possible to use a truncated proposal provided that you modify the MH acceptance ratio to include the relevant normalisation constants. If you don’t modify the acceptance probability, you will get a sampler which targets the wrong distribution, but it will often be quite similar to the correct target, making it a difficult bug to spot and track down.

Or can you transform the target distribution (for example using logs) so that it has full support?
You can if you’re careful, but that can also have issues if there is appreciable mass near zero.
Thanks for the reply and very interesting post. Does your point also apply if you transform the proposal distribution at the same time?
I think the answer is “yes”, but it obviously depends on exactly what you mean. Clearly for the example given you cannot log-transform the proposal, as it has full support.
Nice, clear and to the point as always, Darren. Thanks for this, I have seen many people falling for this one or variants thereof, too.
Two things I would add:
* when using rejection to stay positive, you might get a very long sampling time when proposing from a state close to 0 with a variance “too large” [number of loops needed = geometric distribution with mean $1/(1-CDF(0|x))$ ]
* I would double-check when using this rejection with adaptive MCMC à la Haario, or Rosenthal & Roberts, or Moulines and Andrieu i.e. when learning the poseterior covariance matrix and proposing with a mixture containing a non-adaptive “safety” component. In that case, the impact of the rejection on the learned proposal covariance matrix is not clear to me (in the non-asymptotic case), *plus*, as I presented in a NIPS Workshop last year, rejecting on the outer loop of the mixture will make the weight of the safety component dependent on $x$: have to make sure this is taken into account in the MH ratio — or to condition within each mixture component instead of conditioning the whole mixture.
Nice post, this is how I have been implementing samplers with non-negativity on a couple of occasions.
However, why not use rtnorm from the msm package? It is also useful to vectorize code in hierarchical models.
Thank you so much, your article helped me a lot!
As I understand, the variance parameter in the proposal (normal) distribution, should be adjusted to achieve and acceptance ratio of 30 – 50 % to obtain good mixing. Thus, is it correct that if one draws from a normal(0, V) distribution, the scaling should be the normal cdf of x and xs with mean 0 and variance V?
Useful post. Thank you.
Do the results generalize to the multivariate case? For example, suppose the target distribution includes truncated normal random variables (as well as random variables endowed with inverse gamma distributions) while the proposal remains a Gaussian random walk?
Nice work, thanks. I have two questions about this work. Firstly, why you have used CDF_normal with mean zero and sd=1? Secondly, what about multivariate gaussian proposals?
Nice post. Thanks!
Please can anyone give me R codes for Metropolis, Metropolis-Hastings and Gibbs samplers for sampling from a bivariate normal distribution? I need to compare the methods using different proposal variances. Thank you and it seems rather urgent for me. Thanks again.
as has been pointed out, rejection means waste of computational effort. why don’t you use reflection.
if x* < 0, just set it to -x*, and the proposal ratio is 1, when x*|x is symmetric.
see our paper (Yang, Z. and C. E. Rodríguez (2013). "Searching for efficient Markov chain Monte Carlo proposal kernels." Proc. Natl. Acad. Sci. U.S.A. 110(48): 19307–19312). This also points out that the Gaussian random walk is in general more expensive and less efficient than the simple uniform.
ziheng
i want to ask about implementation metropolis hasting algorithm for Weibull 2-parameters Distribution and numerical simulation with matlab or R? thank’s
Can someone explain how there’s a difference from continuing sampling till you get an all positive result from sampling, getting a negative result, rejecting it, and starting the process all over again. I don’t see the difference.
It took me quite a while to see the difference, and I think it could all be much more obvious if the key point were explained: if you don’t accept the new draw, YOU STAY AT THE OLD ONE. That it, it “counts”, and you get multiple values of it in a row.
It’s easy to verify using MATLAB. Wrong code, mirroring yours:
>> x=1; Draws=[]; for i=1:100000; xs=x+randn(1); y=(xs*exp(-xs))/(x*exp(-x)); if y>rand(1); x=xs; Draws=[Draws; x]; end; end;
>> histogram(Draws, ‘Normalization’, ‘pdf’); hold on; XGamma=[0:.01:10]; YGamma=XGamma.*exp(-XGamma); plot(XGamma, YGamma); hold off;
Correct code, with only the “end” changing places by a bit:
>> x=1; Draws=[]; for i=1:100000; xs=x+randn(1); y=(xs*exp(-xs))/(x*exp(-x)); if y>rand(1); x=xs; end; Draws=[Draws; x]; end;
>> histogram(Draws, ‘Normalization’, ‘pdf’); hold on; XGamma=[0:.01:10]; YGamma=XGamma.*exp(-XGamma); plot(XGamma, YGamma); hold off;
I don’t think the key issue, really, is throwing away the proposals BEFORE calculating the acceptance ratio. It’s simply moving on and not “staying at” the previous value.
If I’m wrong about this, I’d appreciate learning why.
How the constant of proportionality is calculated?
Thanks so much for your post. It helped a lot for my self study.
As a side note, I’m not sure if you are aware that your post has been plagiarized at this blog: http://www.yindawei.com/2014/12/05/metropolis-hastings-mcmc-when-the-proposal-and-target-have-differing-support/
I’m not sure if you care to confront the other blogger, but just want to put a word out there.