Introduction
Parallel tempering is a method for getting Metropolis-Hastings based MCMC algorithms to work better on multi-modal distributions. Although the idea has been around for more than 20 years, and works well on many problems, it still isn’t routinely used in applications. I think this is partly because relatively few people understand how it works, and partly due to the perceived difficulty of implementation. I hope to show here that it is both very easy to understand and to implement. It is also rather easy to implement in parallel on multi-core systems, though I won’t get into that in this post.
Sampling a double-well potential
To illustrate the ideas, we need a toy multi-modal distribution to sample. There are obviously many possibilities here, but I rather like to use a double potential well distribution. The simplest version of this assumes a potential function of the form
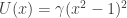
for some given potential barrier height 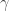
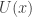
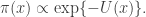
There is a physical explanation for the terminology, via Langevin diffusions, but that isn’t really important here. It is fine to just think of potentials as being a (negative) log-density scale. On this scale, high potential barrier heights correspond to regions of very low probability density. We can set up a double well potential and plot it for the case 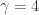
U=function(gam,x)
{
gam*(x*x-1)*(x*x-1)
}
curried=function(gam)
{
message(paste("Returning a function for gamma =",gam))
function(x) U(gam,x)
}
U4=curried(4)
op=par(mfrow=c(2,1))
curve(U4(x),-2,2,main="Potential function, U(x)")
curve(exp(-U4(x)),-2,2,main="Unnormalised density function, exp(-U(x))")
par(op)
leading to the following plot

Incidentally, the function curried(), which curries the potential function, did not include the message() statement when I first wrote it. It mostly worked fine, but some of the code below didn’t behave as I expected. I inserted the message() statement to figure out what was going on, and the code started behaving perfectly – a beautiful example of a Heisenbug! The reason is that the message statement is not redundant – it forces evaluation of the gam variable, which is necessary in some cases, due to the lazy evaluation model that R uses for function arguments. If you don’t like the message() statement, replacing it with a simple gam works just as well.
Anyway, the point is that we have defined a multi-modal density, and that a Metropolis-Hastings algorithm initialised in one of the modes will have a hard time jumping to the other mode, and the difficulty of this jump will increase as we increase the value of
We can write a simple function for a Metropolis algorithm targeting a particular potential function as follows.
chain=function(target,tune=0.1,init=1)
{
x=init
xvec=numeric(iters)
for (i in 1:iters) {
can=x+rnorm(1,0,tune)
logA=target(x)-target(can)
if (log(runif(1))<logA)
x=can
xvec[i]=x
}
xvec
}
We can use this code to run some chains for a few different values of
temps=2^(0:3)
iters=1e5
mat=sapply(lapply(temps,curried),chain)
colnames(mat)=paste("gamma=",temps,sep="")
require(smfsb)
mcmcSummary(mat,rows=length(temps))
leading to the plot below.

We see that as 
Before we see how to couple the chains and improve the mixing, it is useful to think how to re-write this sampler. Above we sampled each chain in turn for different barrier heights. To couple the chains, we need to sample them together, sampling each iteration for all of the chains in turn. This is very easy to do. The code below isn’t especially efficient, but it is written to look very similar to the single chain code above.
chains=function(pot=U, tune=0.1, init=1)
{
x=rep(init,length(temps))
xmat=matrix(0,iters,length(temps))
for (i in 1:iters) {
can=x+rnorm(length(temps),0,tune)
logA=unlist(Map(pot,temps,x))-unlist(Map(pot,temps,can))
accept=(log(runif(length(temps)))<logA)
x[accept]=can[accept]
xmat[i,]=x
}
colnames(xmat)=paste("gamma=",temps,sep="")
xmat
}
mcmcSummary(chains(),rows=length(temps))
This code should behave identically to the previous code, simulating independent parallel MCMC chains. However, the code is now in the form that is very easy to modify to couple the chains together in an attempt to improve mixing.
Coupling parallel chains
In the above example the chains we are simulating are all independent of one another. Some mix reasonably well, and some mix very badly. But the distributions are all related to one another, changing gradually as the barrier height changes. The idea behind coupling the chains is to try and swap states between the chains to use the chains which are mixing well to improve the mixing of the chains which aren’t. In particular, suppose interest is in the target of the worst mixing chain. The other chains can be constructed as “tempered” versions of the target of interest, here by raising it to a power between 0 and 1, with 0 corresponding to a complete flattening of the distribution, and 1 corresponding to the desired target. The use of parallel chains to improve mixing in this way is usually referred to as parallel tempering, but also sometimes as 
So, how do we swap states between the chains without affecting the target distributions? As always, just use a Metropolis-Hastings update… To keep things simple, let’s just suppose that we have two (independent, parallel) chains, one with target 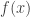



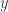
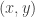
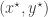



but it really doesn’t matter, as it is clearly a symmetric proposal, and hence will drop out of the M-H ratio. Our acceptance probability is therefore 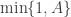

So, if we use this acceptance probability whenever we propose a swap of the states between two chains, then we will preserve the joint target, and hence the marginal targets and asymptotic independence of the target. However, the chains themselves will no longer be independent of one another. They will be coupled – Metropolis coupled. This is very easy to implement. We can just add a few lines to our previous chains() function as follows
chains=function(pot=U, tune=0.1, init=1)
{
x=rep(init,length(temps))
xmat=matrix(0,iters,length(temps))
for (i in 1:iters) {
can=x+rnorm(length(temps),0,tune)
logA=unlist(Map(pot,temps,x))-unlist(Map(pot,temps,can))
accept=(log(runif(length(temps)))<logA)
x[accept]=can[accept]
# now the coupling update
swap=sample(1:length(temps),2)
logA=pot(temps[swap[1]],x[swap[1]])+pot(temps[swap[2]],x[swap[2]])-
pot(temps[swap[1]],x[swap[2]])-pot(temps[swap[2]],x[swap[1]])
if (log(runif(1))<logA)
x[swap]=rev(x[swap])
# end of the coupling update
xmat[i,]=x
}
colnames(xmat)=paste("gamma=",temps,sep="")
xmat
}
This can be used as before, but now gives results as illustrated in the following plots.

We see immediately from the plots that whilst the individual target distributions remain unchanged, the mixing of the chains is greatly improved (though still far from perfect). Note that in the code above I just picked two chains at random to propose a state swap. In practice people typically only propose to swap states between chains which are adjacent – i.e. most similar, since proposed swaps between chains with very different targets are unlikely to be accepted. Since implementation of either strategy is very easy, I would recommend trying both to see which works best.
Parallel implementation
It should be clear that there are opportunities for parallelising the above algorithm to make effective use of modern multi-core hardware. An approach using OpenMP with C++ is discussed in this blog post. Also note that if the state space of the chains is large, it can be much more efficient to swap temperatures between the chains rather than states – so long as you are careful about keeping track of stuff – this is explored in detail in Altekar et al (’04).
References
- G. Altekar et al (2004) Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference, Bioinformatics, 20(3): 407-415.
- C. J. Geyer (2011) Importance sampling, simulated tempering, and umbrella sampling, in the Handbook of Markov Chain Monte Carlo, S. P. Brooks, et al (eds), Chapman & Hall/CRC.
- C. J. Geyer (1991) Markov chain Monte Carlo maximum likelihood, Computing Science and Statistics, 23: 156-163.
Complete R script
For convenience, a complete R script to run all of the examples in this post is given below.
# temper.R
# functions for messing around with tempering MCMC
U=function(gam,x)
{
gam*(x*x-1)*(x*x-1)
}
curried=function(gam)
{
#gam
message(paste("Returning a function for gamma =",gam))
function(x) U(gam,x)
}
U4=curried(4)
op=par(mfrow=c(2,1))
curve(U4(x),-2,2,main="Potential function, U(x)")
curve(exp(-U4(x)),-2,2,main="Unnormalised density function, exp(-U(x))")
par(op)
# global settings
temps=2^(0:3)
iters=1e5
# First look at some independent chains
chain=function(target,tune=0.1,init=1)
{
x=init
xvec=numeric(iters)
for (i in 1:iters) {
can=x+rnorm(1,0,tune)
logA=target(x)-target(can)
if (log(runif(1))<logA)
x=can
xvec[i]=x
}
xvec
}
mat=sapply(lapply(temps,curried),chain)
colnames(mat)=paste("gamma=",temps,sep="")
require(smfsb)
mcmcSummary(mat,rows=length(temps))
# Next, let's generate 5 chains at once...
chains=function(pot=U, tune=0.1, init=1)
{
x=rep(init,length(temps))
xmat=matrix(0,iters,length(temps))
for (i in 1:iters) {
can=x+rnorm(length(temps),0,tune)
logA=unlist(Map(pot,temps,x))-unlist(Map(pot,temps,can))
accept=(log(runif(length(temps)))<logA)
x[accept]=can[accept]
xmat[i,]=x
}
colnames(xmat)=paste("gamma=",temps,sep="")
xmat
}
mcmcSummary(chains(),rows=length(temps))
# Next let's couple the chains...
chains=function(pot=U, tune=0.1, init=1)
{
x=rep(init,length(temps))
xmat=matrix(0,iters,length(temps))
for (i in 1:iters) {
can=x+rnorm(length(temps),0,tune)
logA=unlist(Map(pot,temps,x))-unlist(Map(pot,temps,can))
accept=(log(runif(length(temps)))<logA)
x[accept]=can[accept]
# now the coupling update
swap=sample(1:length(temps),2)
logA=pot(temps[swap[1]],x[swap[1]])+pot(temps[swap[2]],x[swap[2]])-
pot(temps[swap[1]],x[swap[2]])-pot(temps[swap[2]],x[swap[1]])
if (log(runif(1))<logA)
x[swap]=rev(x[swap])
# end of the coupling update
xmat[i,]=x
}
colnames(xmat)=paste("gamma=",temps,sep="")
xmat
}
mcmcSummary(chains(),rows=length(temps))
# eof
